This function imputes genotypes on a population-by-population basis, where populations can be considered panmictic, or imputes the state for presence-absence data.
gl.impute(
x,
method = "neighbour",
fill.residual = TRUE,
parallel = FALSE,
verbose = NULL
)Arguments
- x
Name of the genlight object containing the SNP or presence-absence data [required].
- method
Imputation method, either "frequency" or "HW" or "neighbour" or "random" [default "neighbour"].
- fill.residual
Should any residual missing values remaining after imputation be set to 0, 1, 2 at random, taking into account global allele frequencies at the particular locus [default TRUE].
- parallel
A logical indicating whether multiple cores -if available- should be used for the computations (TRUE), or not (FALSE); requires the package parallel to be installed [default FALSE].
- verbose
Verbosity: 0, silent or fatal errors; 1, begin and end; 2, progress log ; 3, progress and results summary; 5, full report [default 2 or as specified using gl.set.verbosity].
Value
A genlight object with the missing data imputed.
Details
We recommend that imputation be performed on sampling locations, before any aggregation. Imputation is achieved by replacing missing values using either of two methods:
If "frequency", genotypes scored as missing at a locus in an individual are imputed using the average allele frequencies at that locus in the population from which the individual was drawn.
If "HW", genotypes scored as missing at a locus in an individual are imputed by sampling at random assuming Hardy-Weinberg equilibrium. Applies only to genotype data.
If "neighbour", substitute the missing values for the focal individual with the values taken from the nearest neighbour. Repeat with next nearest and so on until all missing values are replaced.
if "random", missing data are substituted by random values (0, 1 or 2).
The nearest neighbour is the one with the smallest Euclidean distance in all the dataset.
The advantage of this approach is that it works regardless of how many individuals are in the population to which the focal individual belongs, and the displacement of the individual is haphazard as opposed to:
(a) Drawing the individual toward the population centroid (HW and Frequency).
(b) Drawing the individual toward the global centroid (glPCA).
Note that loci that are missing for all individuals in a population are not
imputed with method 'frequency' or 'HW'. Consider using the function
gl.filter.allna with by.pop=TRUE to remove them first.
Examples
# \donttest{
require("dartR.data")
# SNP genotype data
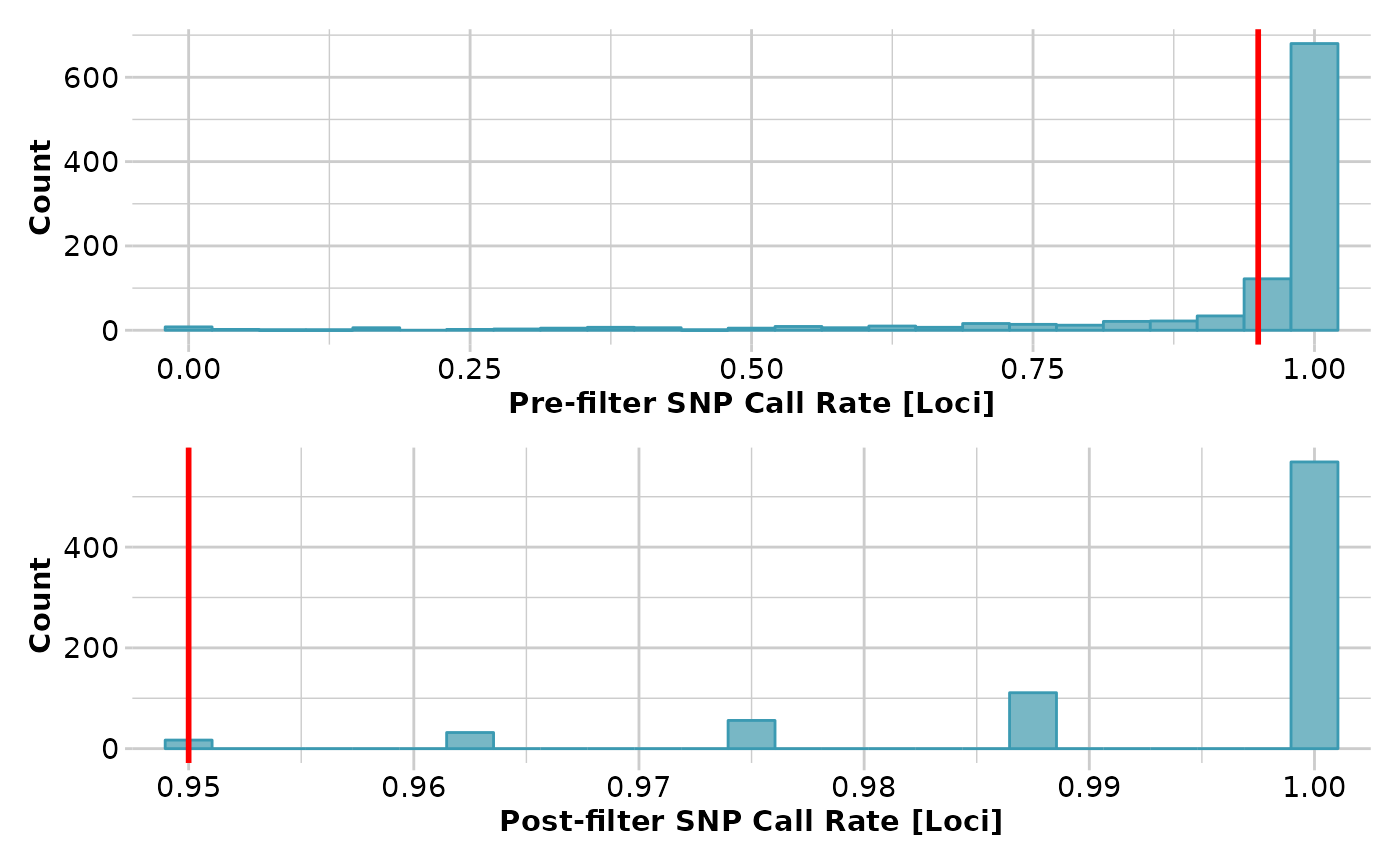
gl <- gl.filter.callrate(platypus.gl,threshold=0.95)
#> Starting gl.filter.callrate
#> Processing genlight object with SNP data
#> Warning: data include loci that are scored NA across all individuals.
#> Consider filtering using gl <- gl.filter.allna(gl)
#> Warning: Data may include monomorphic loci in call rate
#> calculations for filtering
#> Recalculating Call Rate
#> Removing loci based on Call Rate, threshold = 0.95
#>
 #> Completed: gl.filter.callrate
#>
gl <- gl.filter.allna(gl)
#> Starting gl.filter.allna
#> Processing genlight object with SNP data
#> Identifying and removing loci and individuals scored all
#> missing (NA)
#> Deleting loci that are scored as all missing (NA)
#> Deleting individuals that are scored as all missing (NA)
#> Completed: gl.filter.allna
#>
gl <- gl.impute(gl,method="neighbour")
#> Starting gl.impute
#> Processing genlight object with SNP data
#> Imputation based on drawing from the nearest neighbour
#> Residual missing values were filled randomly drawing from the global allele profiles by locus
#> Completed: gl.impute
#>
# Sequence Tag presence-absence data
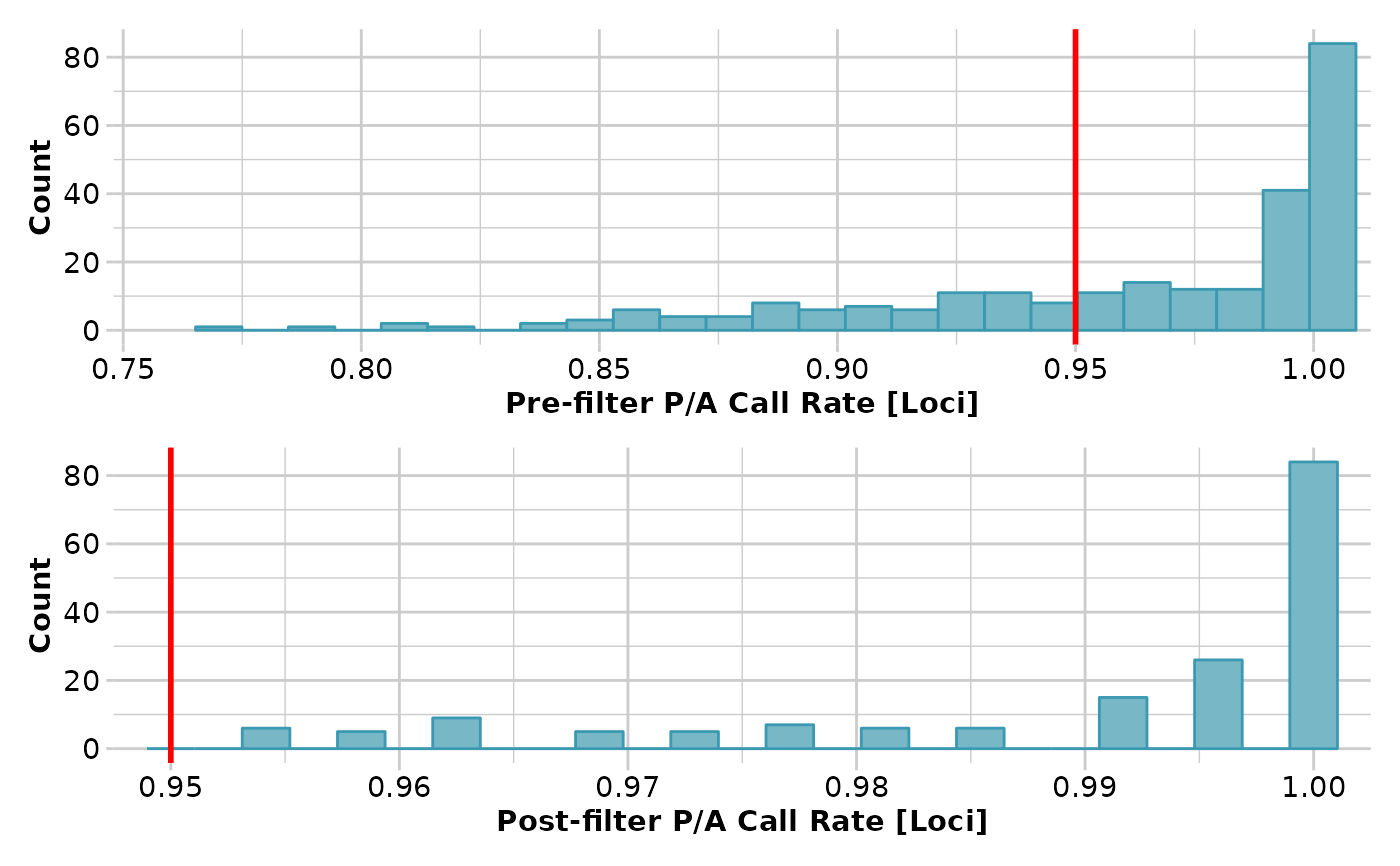
gs <- gl.filter.callrate(testset.gs,threshold=0.95)
#> Starting gl.filter.callrate
#> Processing genlight object with Presence/Absence (SilicoDArT) data
#> Recalculating Call Rate
#> Removing loci based on Call Rate, threshold = 0.95
#>
#> Completed: gl.filter.callrate
#>
gl <- gl.filter.allna(gl)
#> Starting gl.filter.allna
#> Processing genlight object with SNP data
#> Identifying and removing loci and individuals scored all
#> missing (NA)
#> Deleting loci that are scored as all missing (NA)
#> Deleting individuals that are scored as all missing (NA)
#> Completed: gl.filter.allna
#>
gl <- gl.impute(gl,method="neighbour")
#> Starting gl.impute
#> Processing genlight object with SNP data
#> Imputation based on drawing from the nearest neighbour
#> Residual missing values were filled randomly drawing from the global allele profiles by locus
#> Completed: gl.impute
#>
# Sequence Tag presence-absence data
gs <- gl.filter.callrate(testset.gs,threshold=0.95)
#> Starting gl.filter.callrate
#> Processing genlight object with Presence/Absence (SilicoDArT) data
#> Recalculating Call Rate
#> Removing loci based on Call Rate, threshold = 0.95
#>
 #> Completed: gl.filter.callrate
#>
gl <- gl.filter.allna(gl)
#> Starting gl.filter.allna
#> Processing genlight object with SNP data
#> Identifying and removing loci and individuals scored all
#> missing (NA)
#> Deleting loci that are scored as all missing (NA)
#> Deleting individuals that are scored as all missing (NA)
#> Completed: gl.filter.allna
#>
gs <- gl.impute(gs, method="neighbour")
#> Starting gl.impute
#> Processing genlight object with Presence/Absence (SilicoDArT) data
#> Imputation based on drawing from the nearest neighbour
#> Residual missing values were filled randomly drawing from the global allele profiles by locus
#> Completed: gl.impute
#>
# }
gs <- gl.impute(platypus.gl,method ="random")
#> Starting gl.impute
#> Processing genlight object with SNP data
#> Warning: data include loci that are scored NA across all individuals.
#> Consider filtering using gl <- gl.filter.allna(gl)
#> Warning: Population TENTERFIELD has 55 loci with all missing values.
#> Residual missing values were filled randomly drawing from the global allele profiles by locus
#> Completed: gl.impute
#>
#> Completed: gl.filter.callrate
#>
gl <- gl.filter.allna(gl)
#> Starting gl.filter.allna
#> Processing genlight object with SNP data
#> Identifying and removing loci and individuals scored all
#> missing (NA)
#> Deleting loci that are scored as all missing (NA)
#> Deleting individuals that are scored as all missing (NA)
#> Completed: gl.filter.allna
#>
gs <- gl.impute(gs, method="neighbour")
#> Starting gl.impute
#> Processing genlight object with Presence/Absence (SilicoDArT) data
#> Imputation based on drawing from the nearest neighbour
#> Residual missing values were filled randomly drawing from the global allele profiles by locus
#> Completed: gl.impute
#>
# }
gs <- gl.impute(platypus.gl,method ="random")
#> Starting gl.impute
#> Processing genlight object with SNP data
#> Warning: data include loci that are scored NA across all individuals.
#> Consider filtering using gl <- gl.filter.allna(gl)
#> Warning: Population TENTERFIELD has 55 loci with all missing values.
#> Residual missing values were filled randomly drawing from the global allele profiles by locus
#> Completed: gl.impute
#>