Filters loci based on counts of sequence tags scored at a locus (read depth)
Source:R/gl.filter.rdepth.r
gl.filter.rdepth.RdSNP datasets generated by DArT report AvgCountRef and AvgCountSnp as counts of sequence tags for the reference and alternate alleles respectively. These can be used to back calculate Read Depth. Fragment presence/absence datasets as provided by DArT (SilicoDArT) provide Average Read Depth and Standard Deviation of Read Depth as standard columns in their report.
Filtering on Read Depth using the companion script gl.filter.rdepth can be on the basis of loci with exceptionally low counts, or loci with exceptionally high counts.
gl.filter.rdepth(
x,
lower = 5,
upper = 50,
plot.out = TRUE,
plot_theme = theme_dartR(),
plot_colors = two_colors,
save2tmp = FALSE,
verbose = NULL
)Arguments
- x
Name of the genlight object containing the SNP or tag presence/absence data [required].
- lower
Lower threshold value below which loci will be removed [default 5].
- upper
Upper threshold value above which loci will be removed [default 50].
- plot.out
Specify if plot is to be produced [default TRUE].
- plot_theme
Theme for the plot. See Details for options [default theme_dartR()].
- plot_colors
List of two color names for the borders and fill of the plots [default two_colors].
- save2tmp
If TRUE, saves any ggplots and listings to the session temporary directory (tempdir) [default FALSE].
- verbose
Verbosity: 0, silent or fatal errors; 1, begin and end; 2, progress log; 3, progress and results summary; 5, full report [default 2, unless specified using gl.set.verbosity].
Value
Returns a genlight object retaining loci with a Read Depth in the range specified by the lower and upper threshold.
Details
For examples of themes, see:
See also
gl.filter.rdepth
Other filter functions:
gl.filter.allna(),
gl.filter.callrate(),
gl.filter.heterozygosity(),
gl.filter.hwe(),
gl.filter.locmetric(),
gl.filter.maf(),
gl.filter.monomorphs(),
gl.filter.overshoot(),
gl.filter.pa(),
gl.filter.parent.offspring(),
gl.filter.reproducibility(),
gl.filter.secondaries(),
gl.filter.sexlinked(),
gl.filter.taglength()
Examples
# \donttest{
# SNP data
gl.report.rdepth(testset.gl)
#> Starting gl.report.rdepth
#> Processing genlight object with SNP data
#> Reporting Read Depth by Locus
#> No. of loci = 255
#> No. of individuals = 250
#> Minimum : 3.1
#> 1st quartile : 6.3
#> Median : 9.5
#> Mean : 16.60392
#> 3r quartile : 22.75
#> Maximum : 66.4
#> Missing Rate Overall: 0.12
#>
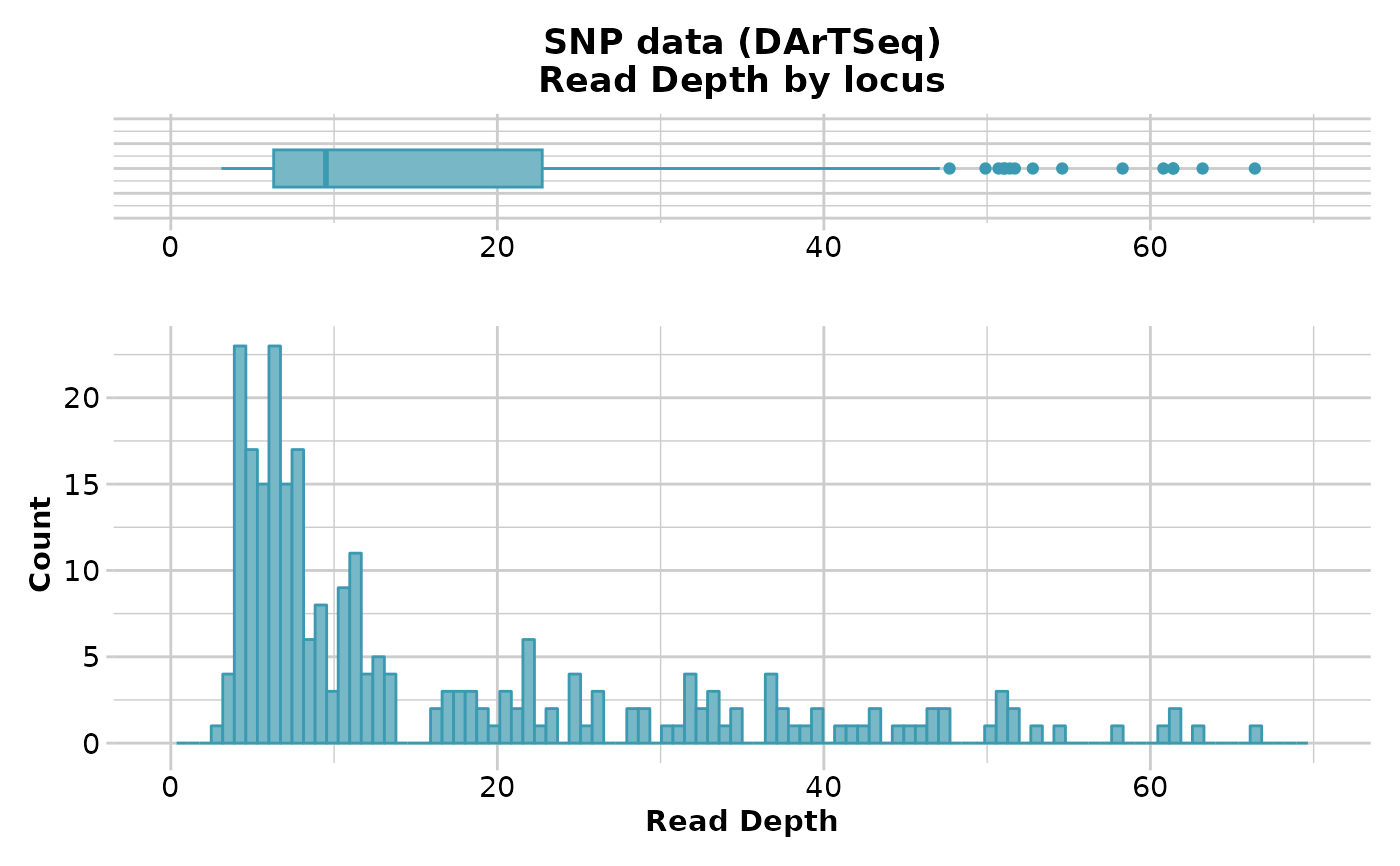 #> Quantile Threshold Retained Percent Filtered Percent
#> 1 100% 66.4 1 0.4 254 99.6
#> 2 95% 50.7 13 5.1 242 94.9
#> 3 90% 41.0 26 10.2 229 89.8
#> 4 85% 34.2 39 15.3 216 84.7
#> 5 80% 28.9 52 20.4 203 79.6
#> 6 75% 23.0 64 25.1 191 74.9
#> 7 70% 20.0 77 30.2 178 69.8
#> 8 65% 16.2 90 35.3 165 64.7
#> 9 60% 12.1 102 40.0 153 60.0
#> 10 55% 10.8 116 45.5 139 54.5
#> 11 50% 9.5 129 50.6 126 49.4
#> 12 45% 8.1 143 56.1 112 43.9
#> 13 40% 7.5 157 61.6 98 38.4
#> 14 35% 7.0 167 65.5 88 34.5
#> 15 30% 6.5 182 71.4 73 28.6
#> 16 25% 6.3 192 75.3 63 24.7
#> 17 20% 5.6 205 80.4 50 19.6
#> 18 15% 4.9 217 85.1 38 14.9
#> 19 10% 4.4 232 91.0 23 9.0
#> 20 5% 4.2 244 95.7 11 4.3
#> 21 0% 3.1 255 100.0 0 0.0
#> Completed: gl.report.rdepth
#>
result <- gl.filter.rdepth(testset.gl, lower=8, upper=50, verbose=3)
#> Starting gl.filter.rdepth
#> Processing genlight object with SNP data
#> Removing loci with rdepth <= 8 and >= 50
#>
#> Quantile Threshold Retained Percent Filtered Percent
#> 1 100% 66.4 1 0.4 254 99.6
#> 2 95% 50.7 13 5.1 242 94.9
#> 3 90% 41.0 26 10.2 229 89.8
#> 4 85% 34.2 39 15.3 216 84.7
#> 5 80% 28.9 52 20.4 203 79.6
#> 6 75% 23.0 64 25.1 191 74.9
#> 7 70% 20.0 77 30.2 178 69.8
#> 8 65% 16.2 90 35.3 165 64.7
#> 9 60% 12.1 102 40.0 153 60.0
#> 10 55% 10.8 116 45.5 139 54.5
#> 11 50% 9.5 129 50.6 126 49.4
#> 12 45% 8.1 143 56.1 112 43.9
#> 13 40% 7.5 157 61.6 98 38.4
#> 14 35% 7.0 167 65.5 88 34.5
#> 15 30% 6.5 182 71.4 73 28.6
#> 16 25% 6.3 192 75.3 63 24.7
#> 17 20% 5.6 205 80.4 50 19.6
#> 18 15% 4.9 217 85.1 38 14.9
#> 19 10% 4.4 232 91.0 23 9.0
#> 20 5% 4.2 244 95.7 11 4.3
#> 21 0% 3.1 255 100.0 0 0.0
#> Completed: gl.report.rdepth
#>
result <- gl.filter.rdepth(testset.gl, lower=8, upper=50, verbose=3)
#> Starting gl.filter.rdepth
#> Processing genlight object with SNP data
#> Removing loci with rdepth <= 8 and >= 50
#>
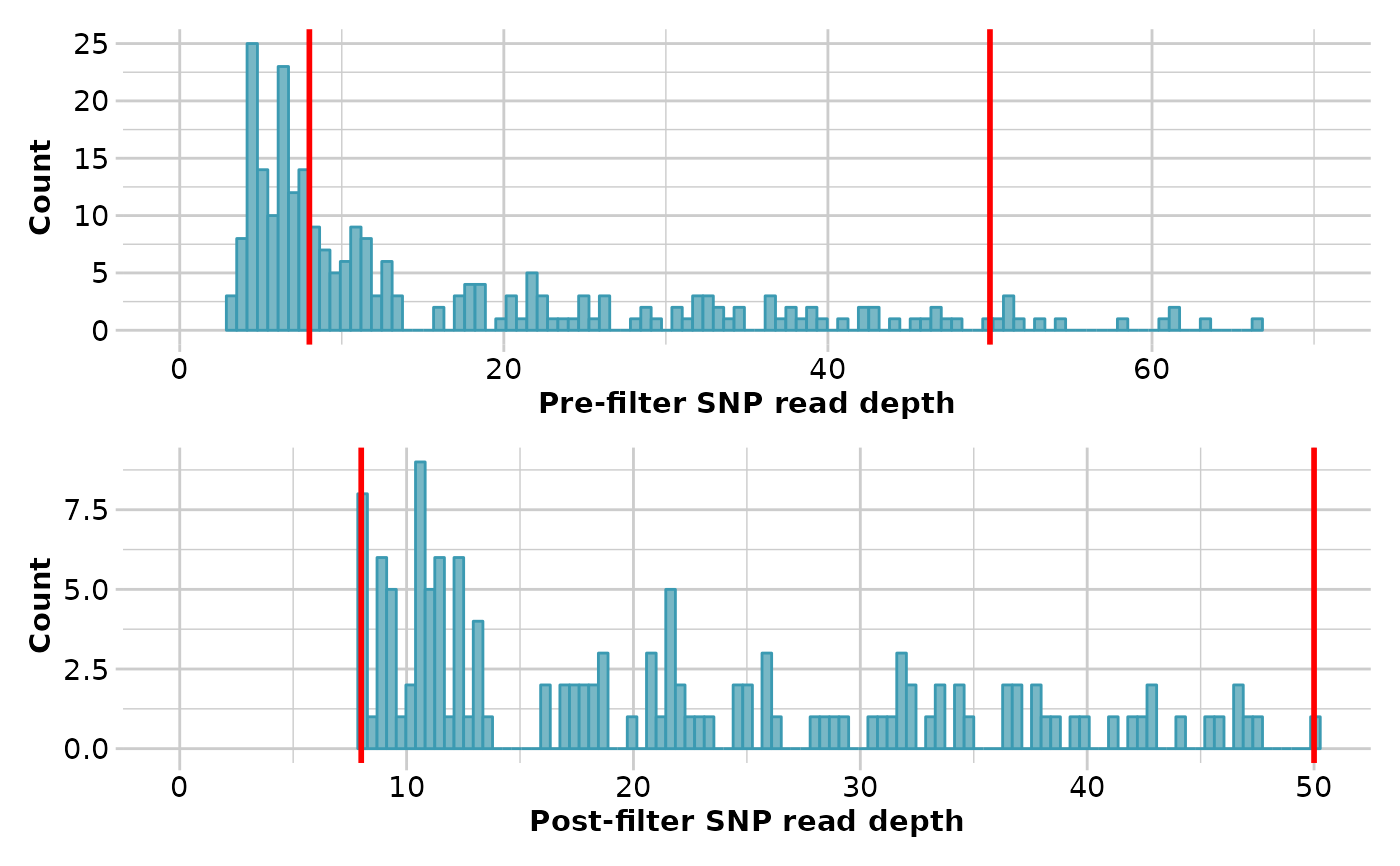 #> Summary of filtered dataset
#> Initial no. of loci = 255
#> No. of loci deleted = 122
#> No. of loci retained: 133
#> No. of individuals: 250
#> No. of populations: 30
#> Completed: gl.filter.rdepth
#>
# Tag P/A data
result <- gl.filter.rdepth(testset.gs, lower=8, upper=50, verbose=3)
#> Starting gl.filter.rdepth
#> Processing genlight object with Presence/Absence (SilicoDArT) data
#> Removing loci with rdepth <= 8 and >= 50
#>
#> Summary of filtered dataset
#> Initial no. of loci = 255
#> No. of loci deleted = 122
#> No. of loci retained: 133
#> No. of individuals: 250
#> No. of populations: 30
#> Completed: gl.filter.rdepth
#>
# Tag P/A data
result <- gl.filter.rdepth(testset.gs, lower=8, upper=50, verbose=3)
#> Starting gl.filter.rdepth
#> Processing genlight object with Presence/Absence (SilicoDArT) data
#> Removing loci with rdepth <= 8 and >= 50
#>
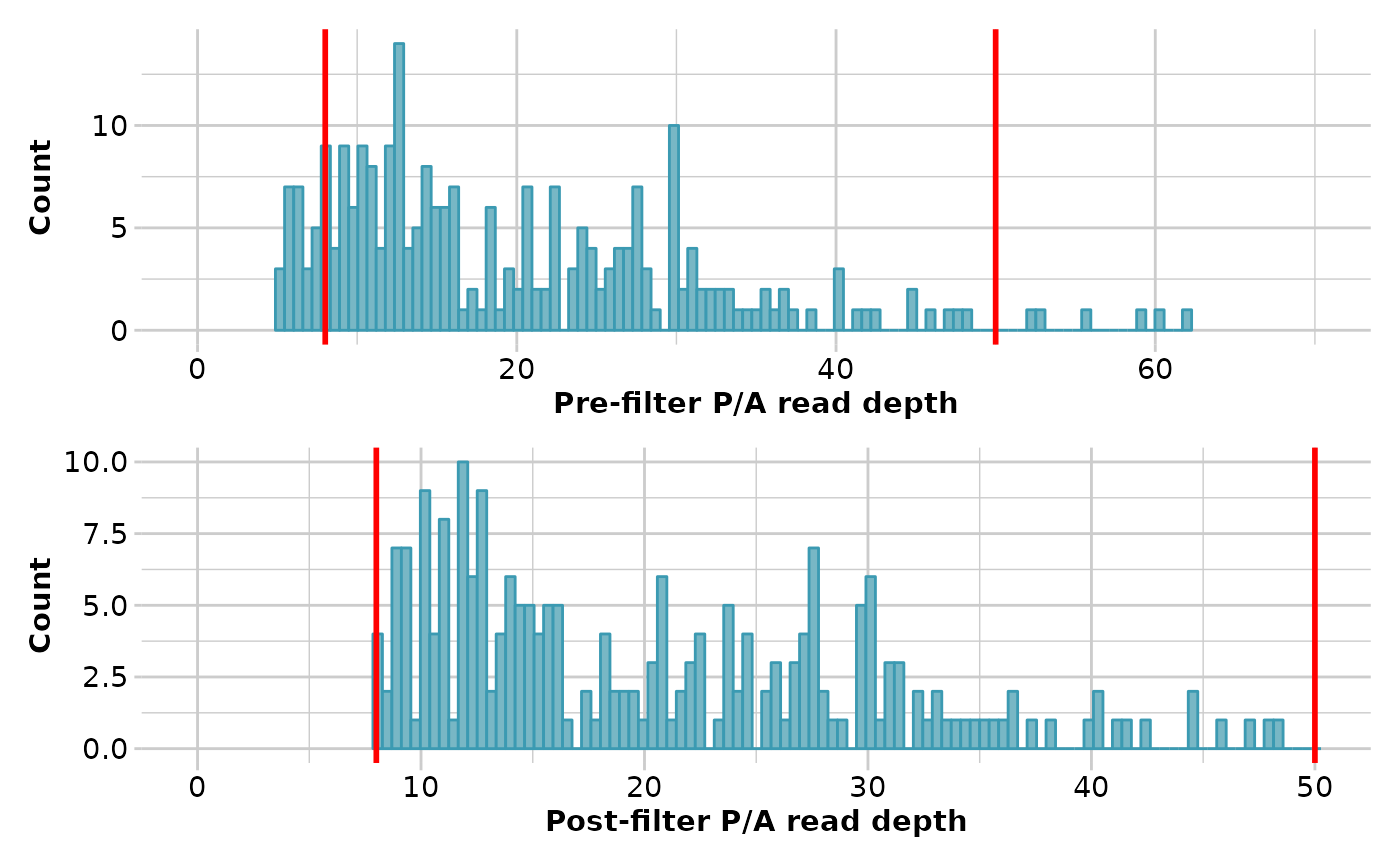 #> Summary of filtered dataset
#> Initial no. of loci = 255
#> No. of loci deleted = 35
#> No. of loci retained: 220
#> No. of individuals: 218
#> No. of populations: 29
#> Completed: gl.filter.rdepth
#>
# }
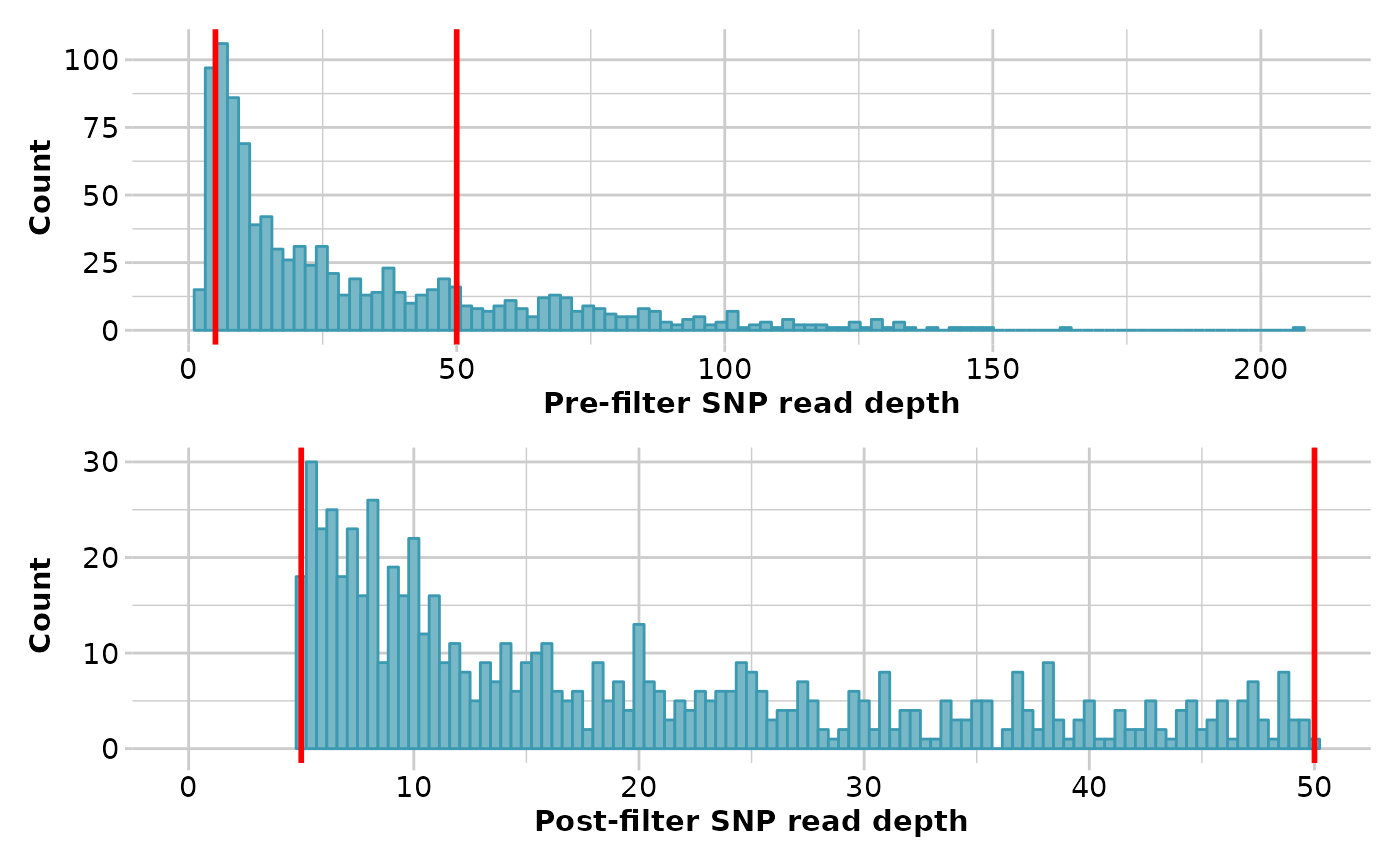
res <- gl.filter.rdepth(platypus.gl)
#> Starting gl.filter.rdepth
#> Processing genlight object with SNP data
#> Warning: data include loci that are scored NA across all individuals.
#> Consider filtering using gl <- gl.filter.allna(gl)
#> Removing loci with rdepth <= 5 and >= 50
#>
#> Summary of filtered dataset
#> Initial no. of loci = 255
#> No. of loci deleted = 35
#> No. of loci retained: 220
#> No. of individuals: 218
#> No. of populations: 29
#> Completed: gl.filter.rdepth
#>
# }
res <- gl.filter.rdepth(platypus.gl)
#> Starting gl.filter.rdepth
#> Processing genlight object with SNP data
#> Warning: data include loci that are scored NA across all individuals.
#> Consider filtering using gl <- gl.filter.allna(gl)
#> Removing loci with rdepth <= 5 and >= 50
#>
 #> Completed: gl.filter.rdepth
#>
#> Completed: gl.filter.rdepth
#>