Identifies putative parent offspring within a population
Source:R/gl.report.parent.offspring.r
gl.report.parent.offspring.RdThis script examines the frequency of pedigree inconsistent loci, that is, those loci that are homozygotes in the parent for the reference allele, and homozygous in the offspring for the alternate allele. This condition is not consistent with any pedigree, regardless of the (unknown) genotype of the other parent. The pedigree inconsistent loci are counted as an indication of whether or not it is reasonable to propose the two individuals are in a parent-offspring relationship.
gl.report.parent.offspring(
x,
min.rdepth = 12,
min.reproducibility = 1,
range = 1.5,
plot.out = TRUE,
plot_theme = theme_dartR(),
plot_colors = two_colors,
save2tmp = FALSE,
verbose = NULL
)Arguments
- x
Name of the genlight object containing the SNP genotypes [required].
- min.rdepth
Minimum read depth to include in analysis [default 12].
- min.reproducibility
Minimum reproducibility to include in analysis [default 1].
- range
Specifies the range to extend beyond the interquartile range for delimiting outliers [default 1.5 interquartile ranges].
- plot.out
Creates a plot that shows the sex linked markers [default TRUE].
- plot_theme
Theme for the plot. See Details for options [default theme_dartR()].
- plot_colors
List of two color names for the borders and fill of the plots [default two_colors].
- save2tmp
If TRUE, saves any ggplots and listings to the session temporary directory (tempdir) [default FALSE].
- verbose
Verbosity: 0, silent or fatal errors; 1, begin and end; 2, progress log; 3, progress and results summary; 5, full report [default 2, unless specified using gl.set.verbosity].
Value
A set of individuals in parent-offspring relationship. NULL if no parent-offspring relationships were found.
Details
If two individuals are in a parent offspring relationship, the true number of pedigree inconsistent loci should be zero, but SNP calling is not infallible. Some loci will be miss-called. The problem thus becomes one of determining if the two focal individuals have a count of pedigree inconsistent loci less than would be expected of typical unrelated individuals. There are some quite sophisticated software packages available to formally apply likelihoods to the decision, but we use a simple outlier comparison.
To reduce the frequency of miss-calls, and so emphasize the difference between true parent-offspring pairs and unrelated pairs, the data can be filtered on read depth.
Typically minimum read depth is set to 5x, but you can examine the
distribution of read depths with the function gl.report.rdepth
and push this up with an acceptable loss of loci. 12x might be a good minimum
for this particular analysis. It is sensible also to push the minimum
reproducibility up to 1, if that does not result in an unacceptable loss of
loci. Reproducibility is stored in the slot @other$loc.metrics$RepAvg
and is defined as the proportion of technical replicate assay pairs for which
the marker score is consistent. You can examine the distribution of
reproducibility with the function gl.report.reproducibility.
Note that the null expectation is not well defined, and the power reduced, if the population from which the putative parent-offspring pairs are drawn contains many sibs. Note also that if an individual has been genotyped twice in the dataset, the replicate pair will be assessed by this script as being in a parent-offspring relationship.
The function gl.filter.parent.offspring will filter out those
individuals in a parent offspring relationship.
Note that if your dataset does not contain RepAvg or rdepth among the locus metrics, the filters for reproducibility and read depth are no used.
Function's output
Plots and table are saved to the temporal directory (tempdir) and can be
accessed with the function gl.print.reports and listed with
the function gl.list.reports. Note that they can be accessed
only in the current R session because tempdir is cleared each time that the
R session is closed.
Examples of other themes that can be used can be consulted in
See also
gl.list.reports, gl.report.rdepth ,
gl.print.reports,gl.report.reproducibility,
gl.filter.parent.offspring
Other report functions:
gl.report.bases(),
gl.report.callrate(),
gl.report.diversity(),
gl.report.hamming(),
gl.report.hwe(),
gl.report.ld.map(),
gl.report.locmetric(),
gl.report.maf(),
gl.report.monomorphs(),
gl.report.overshoot(),
gl.report.pa(),
gl.report.rdepth(),
gl.report.replicates(),
gl.report.reproducibility(),
gl.report.secondaries(),
gl.report.sexlinked(),
gl.report.taglength()
Examples
out <- gl.report.parent.offspring(testset.gl[1:10,1:100])
#> Starting gl.report.parent.offspring
#> Processing genlight object with SNP data
#> Warning: data include loci that are scored NA across all individuals.
#> Consider filtering using gl <- gl.filter.allna(gl)
#> Generating null expectation for distribution of counts of
#> pedigree incompatibility
#>
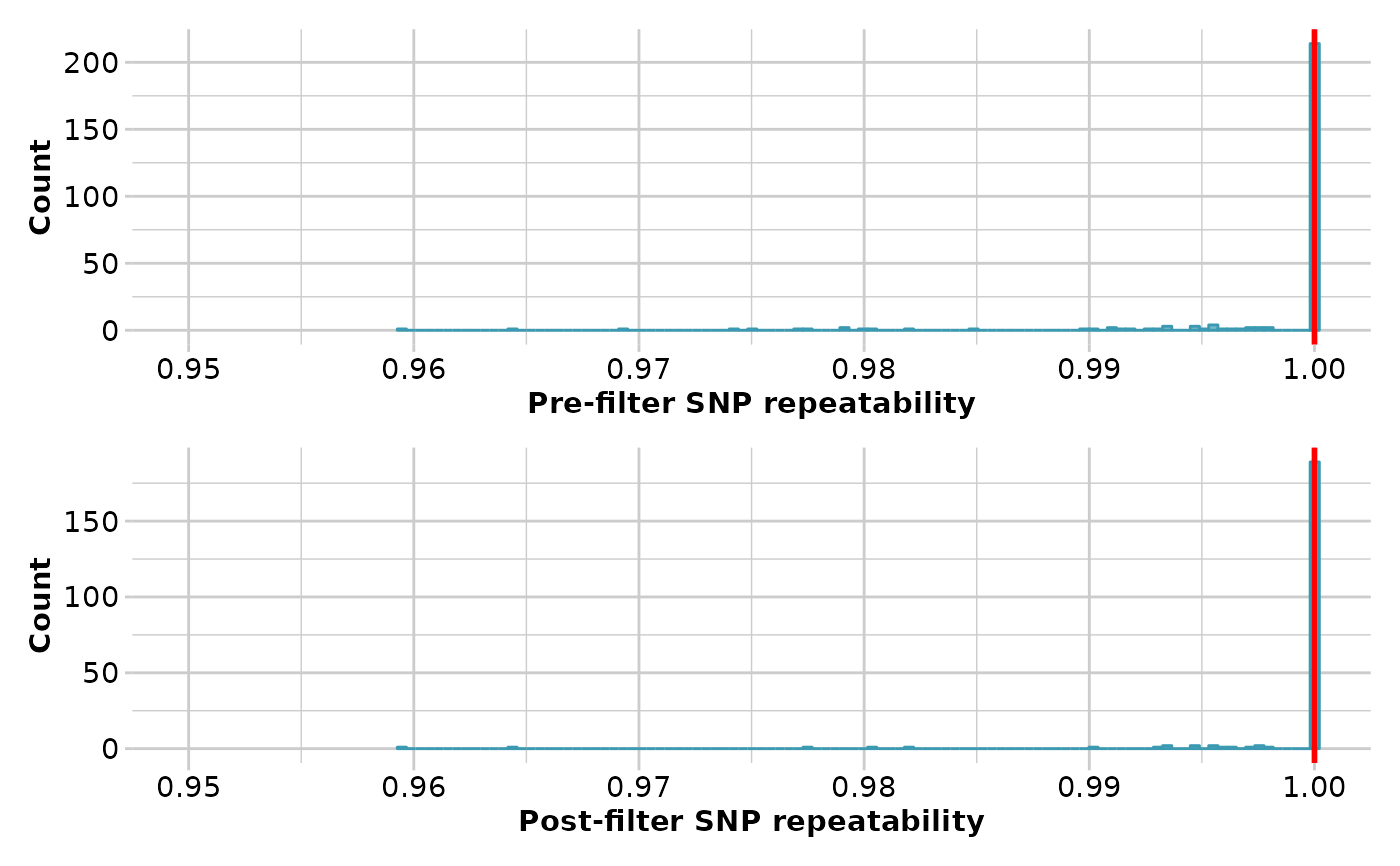
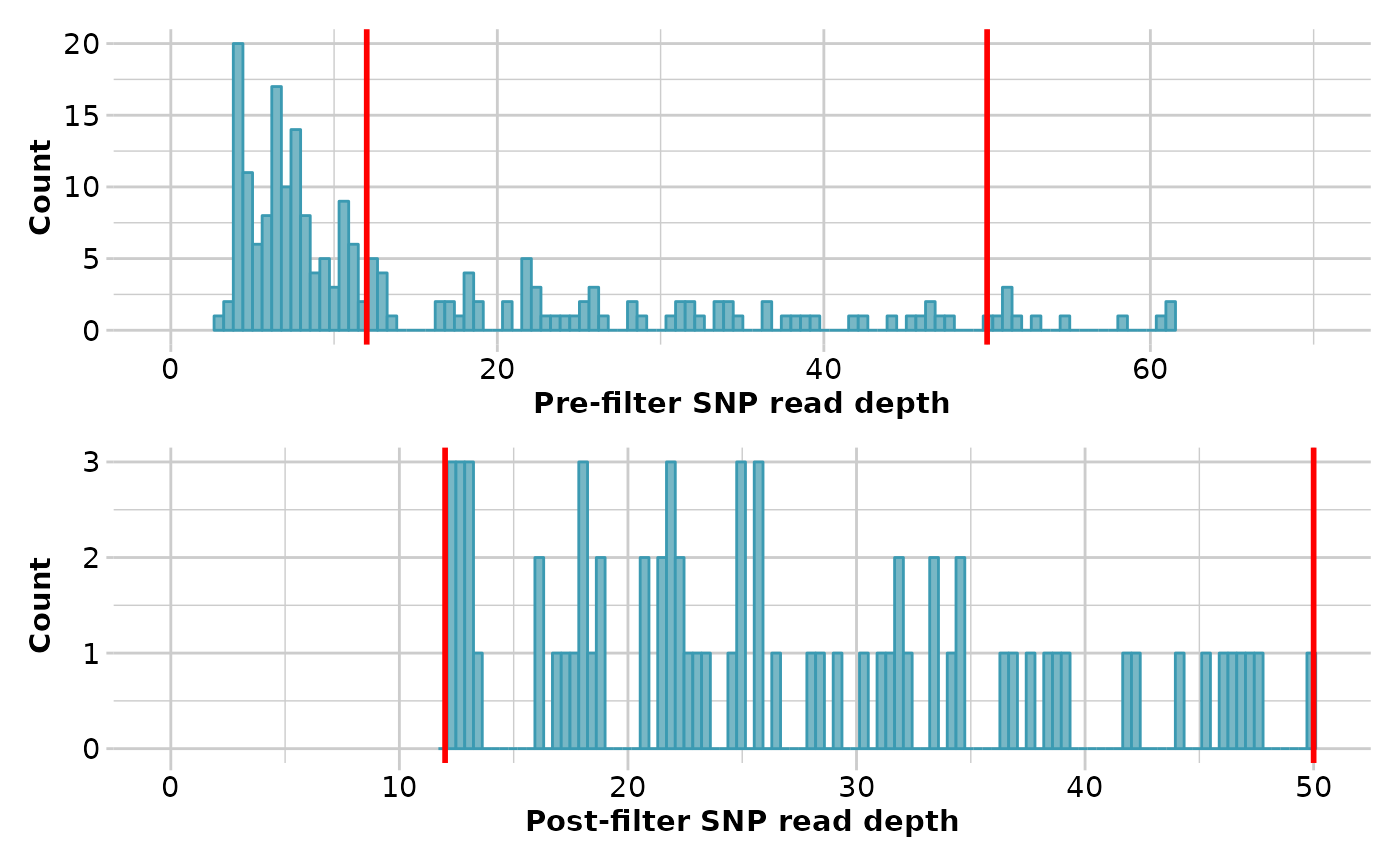 #> Identifying outliers with lower than expected counts of
#> pedigree inconsistencies
#> Identifying outlying pairs
#> No outliers detected
#>
#> Identifying outliers with lower than expected counts of
#> pedigree inconsistencies
#> Identifying outlying pairs
#> No outliers detected
#>
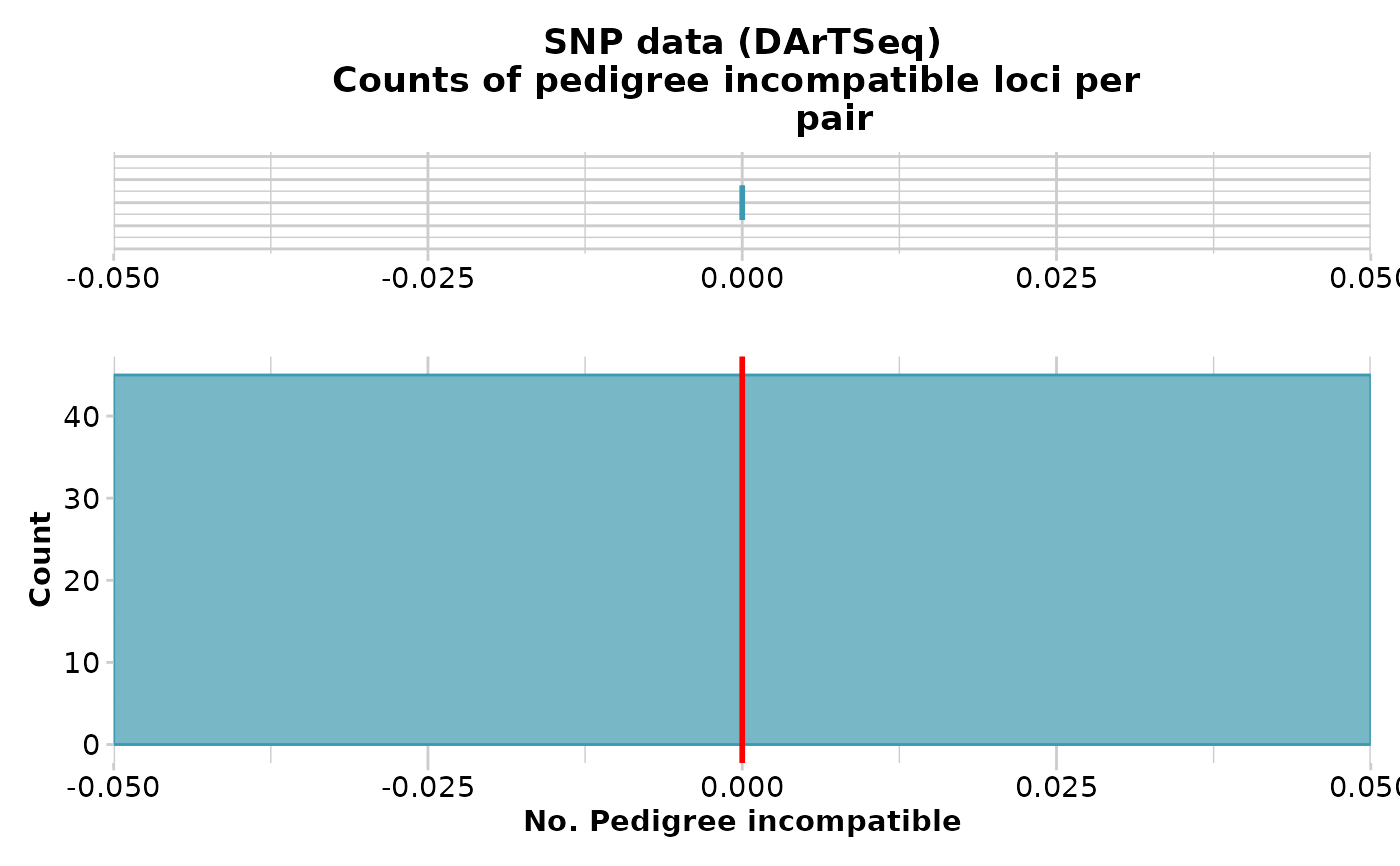 #> Completed: gl.report.parent.offspring
#>
#> Completed: gl.report.parent.offspring
#>