library(dartR.base)
library(dartR.sexlinked)10 Sex Linked Markers
Session presenters

Required packages
Dataset 1 - ZW//ZZ - The Yellow Tufted Honeyeater

Load data
#data("YTH")
load('./data/YTH.rda')
YTH # Explore the dataset ********************
*** DARTR OBJECT ***
********************
** 609 genotypes, 994 SNPs , size: 49.9 Mb
missing data: 139174 (=22.99 %) scored as NA
** Genetic data
@gen: list of 609 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 609 individual labels
@loc.names: 994 locus labels
@loc.all: 994 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 12-516)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, pop, sex, sex_original, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Lichenostomus_HeHo_v1, ChromPos_Lichenostomus_HeHo_v1, AlnCnt_Lichenostomus_HeHo_v1, AlnEvalue_Lichenostomus_HeHo_v1, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, maf
@other$latlon[g]: no coordinates attachedYTH@n.loc # Number of SNPs[1] 994length(YTH@ind.names) # Number of individuals[1] 609Run gl.filter.sexlinked
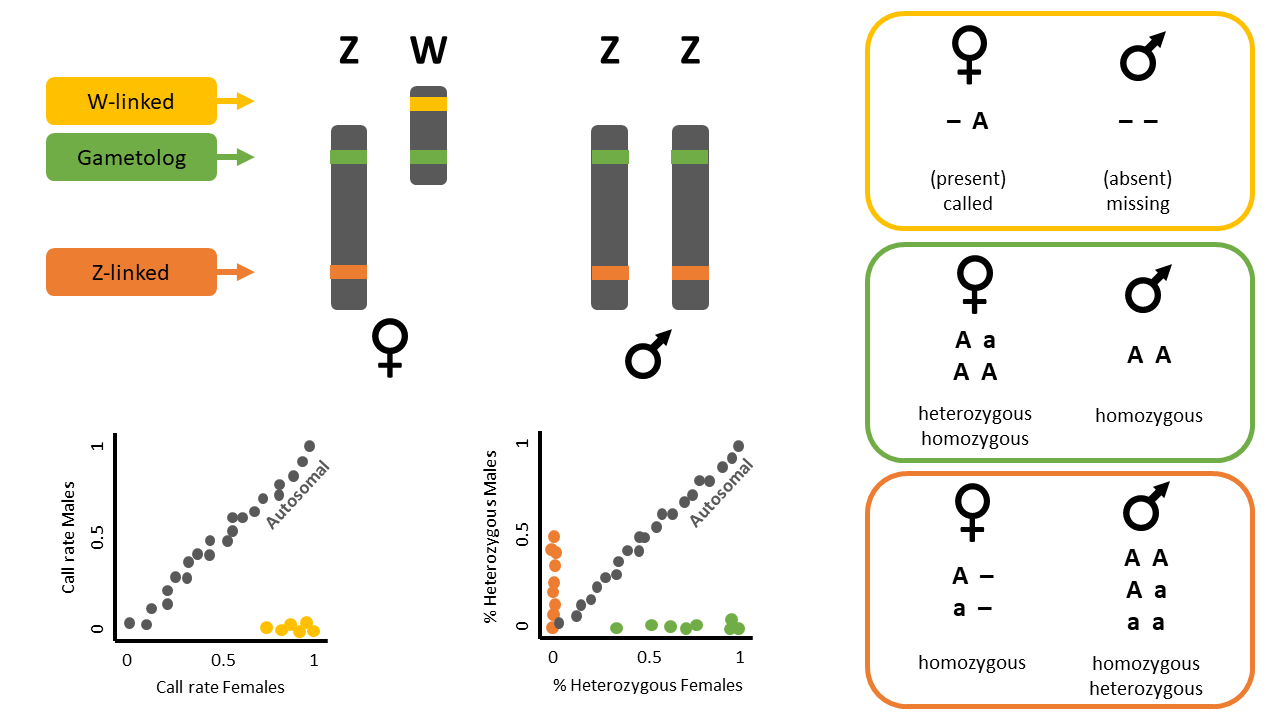
This function identifies sex-linked and autosomal loci present in a SNP dataset (i.e., genlight object) using individuals with known sex. It identifies five types of loci: w-linked or y-linked, sex-biased, z-linked or x-linked, gametologous and autosomal.
The genlight object must contain in gl@other$ind.metrics a column named “id”, and a column named “sex” in which individuals with known-sex are assigned ‘M’ for male, or ‘F’ for female. The function ignores individuals that are assigned anything else or nothing at all (unknown-sex).
Caution
NOTE
Set ncores to more than 1 (default) if you have more than 50,000 SNPs, since it could actually slow down the analysis with smaller datasets.
knitr::kable(head(YTH@other$ind.metrics)) # Check that ind.metrics has the necessary columns| id | pop | sex | sex_original | service | plate_location | |
|---|---|---|---|---|---|---|
| ANWC46839 | ANWC46839 | Melanops | F | F | DLich17-2918 | 1-A1 |
| W49 | W49 | Cassidix | F | F | DLich17-2918 | 1-A10 |
| W90 | W90 | Cassidix | F | F | DLich17-2918 | 1-A12 |
| C25 | C25 | Cassidix | M | M | DLich17-2918 | 1-A2 |
| C8 | C8 | Cassidix | M | M | DLich17-2918 | 1-A3 |
| W70 | W70 | Cassidix | F | F | DLich17-2918 | 1-A4 |
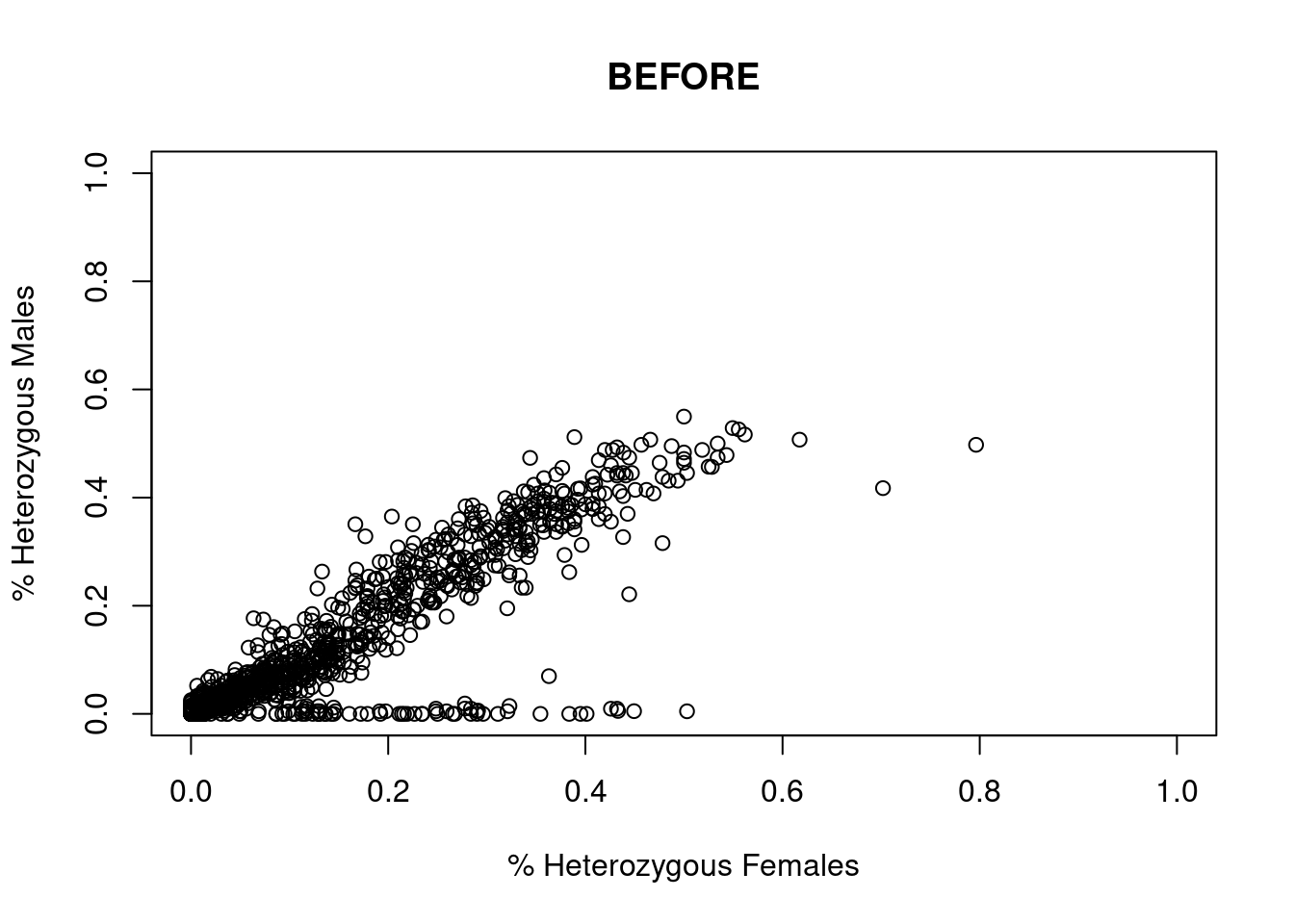
res <- dartR.sexlinked::gl.filter.sexlinked(gl = YTH, system = "zw")Detected 276 females and 333 males.Starting phase 1. May take a while...Building call rate plots.
Done. Starting phase 2.Building heterozygosity plots.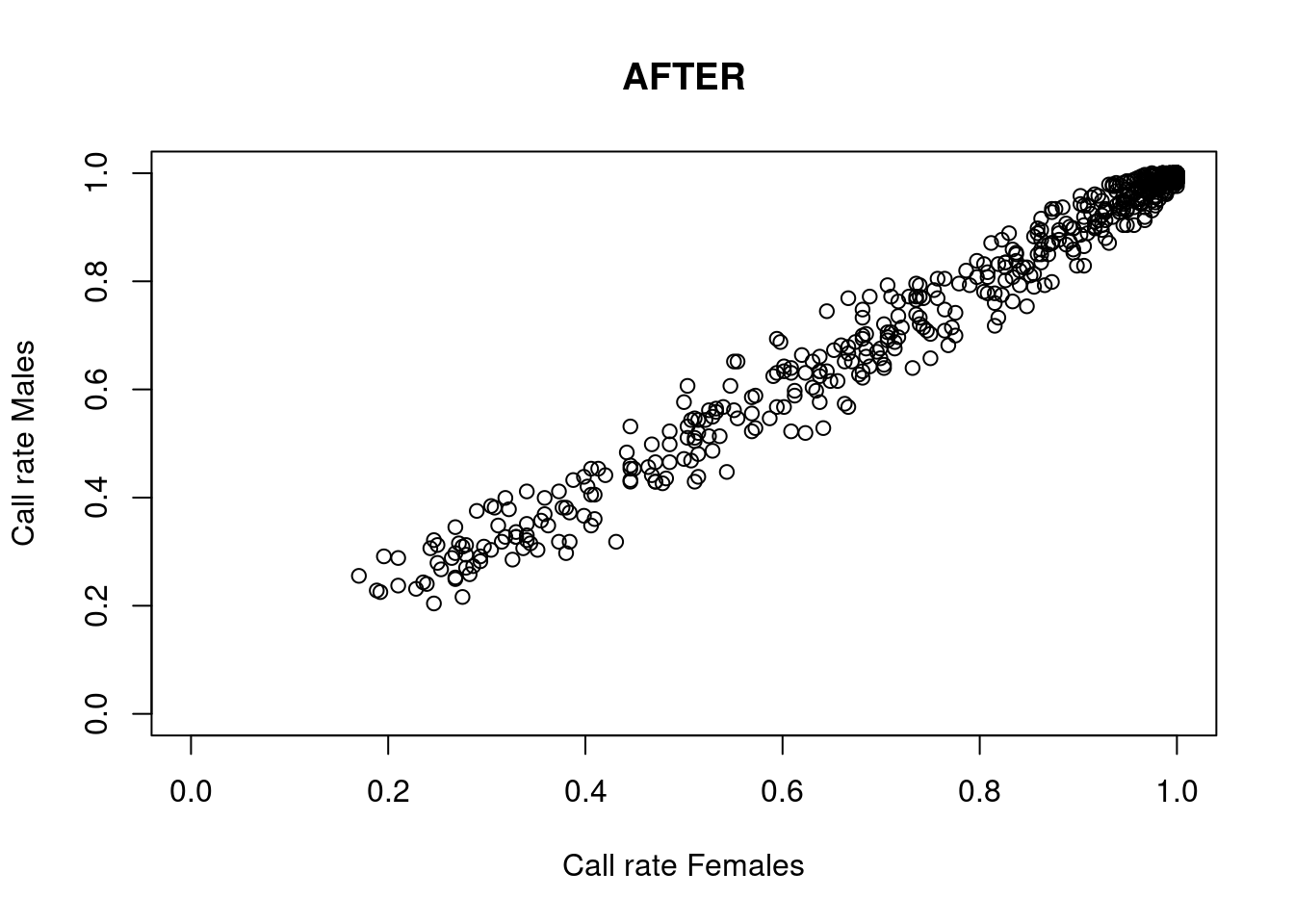

Done building heterozygosity plots.**FINISHED** Total of analyzed loci: 994.
Found 506 sex-linked loci:
52 W-linked loci
273 sex-biased loci
165 Z-linked loci
16 ZW gametologs.
And 488 autosomal loci.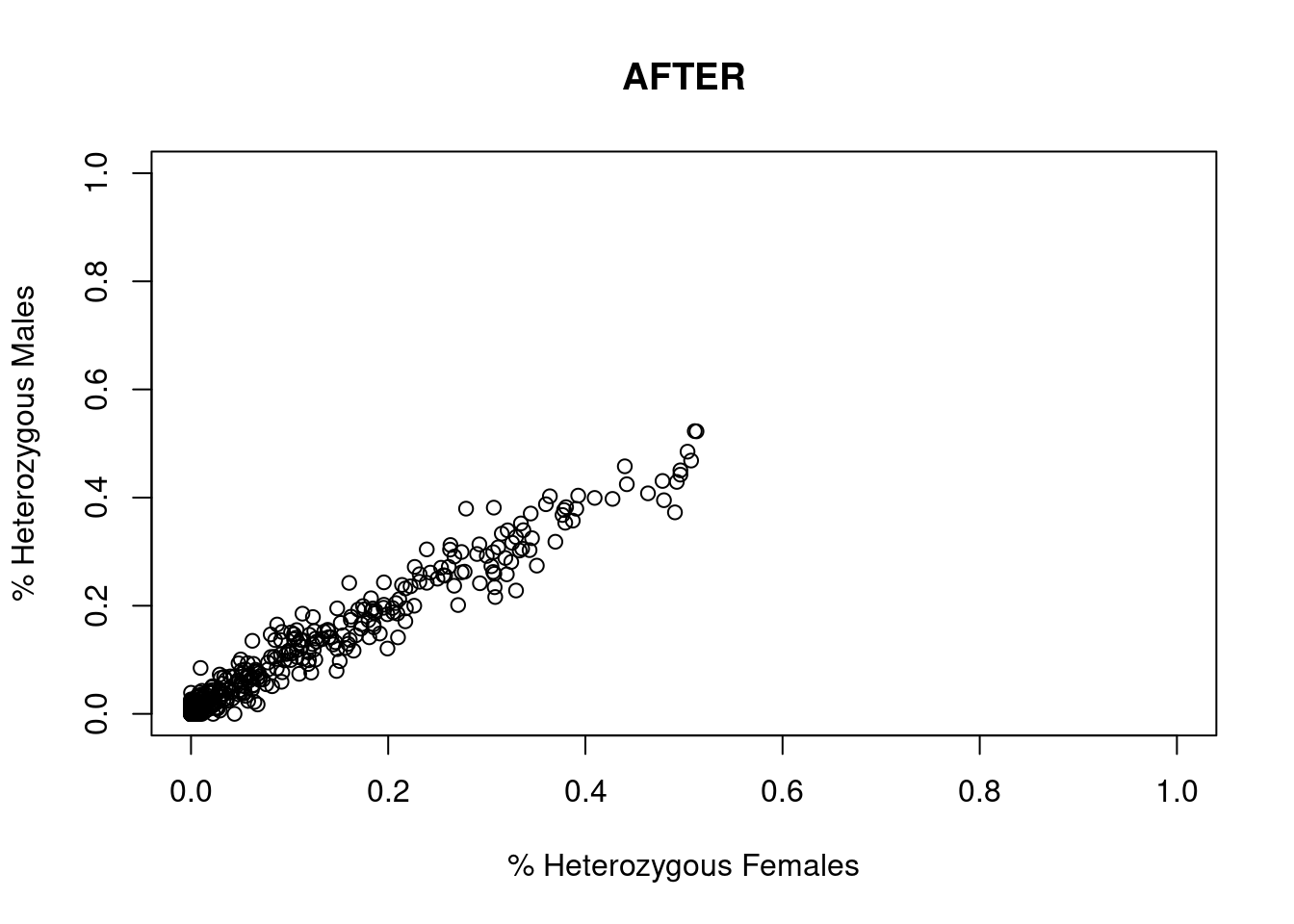
Exercise
![]()
How many males and females does the dataset contain?
How many sex-linked loci were found?
Now check the output:
res$w.linked # Notice that it says "w-linked" ********************
*** DARTR OBJECT ***
********************
** 609 genotypes, 52 SNPs , size: 48.9 Mb
missing data: 17304 (=54.64 %) scored as NA
** Genetic data
@gen: list of 609 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 609 individual labels
@loc.names: 52 locus labels
@loc.all: 52 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 12-516)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, pop, sex, sex_original, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Lichenostomus_HeHo_v1, ChromPos_Lichenostomus_HeHo_v1, AlnCnt_Lichenostomus_HeHo_v1, AlnEvalue_Lichenostomus_HeHo_v1, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, maf
@other$latlon[g]: no coordinates attachedres$z.linked # Notice that it says "z-linked" ********************
*** DARTR OBJECT ***
********************
** 609 genotypes, 165 SNPs , size: 48.9 Mb
missing data: 2990 (=2.98 %) scored as NA
** Genetic data
@gen: list of 609 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 609 individual labels
@loc.names: 165 locus labels
@loc.all: 165 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 12-516)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, pop, sex, sex_original, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Lichenostomus_HeHo_v1, ChromPos_Lichenostomus_HeHo_v1, AlnCnt_Lichenostomus_HeHo_v1, AlnEvalue_Lichenostomus_HeHo_v1, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, maf
@other$latlon[g]: no coordinates attachedres$gametolog ********************
*** DARTR OBJECT ***
********************
** 609 genotypes, 16 SNPs , size: 48.8 Mb
missing data: 580 (=5.95 %) scored as NA
** Genetic data
@gen: list of 609 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 609 individual labels
@loc.names: 16 locus labels
@loc.all: 16 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 12-516)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, pop, sex, sex_original, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Lichenostomus_HeHo_v1, ChromPos_Lichenostomus_HeHo_v1, AlnCnt_Lichenostomus_HeHo_v1, AlnEvalue_Lichenostomus_HeHo_v1, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, maf
@other$latlon[g]: no coordinates attachedres$sex.biased ********************
*** DARTR OBJECT ***
********************
** 609 genotypes, 273 SNPs , size: 49.2 Mb
missing data: 46048 (=27.7 %) scored as NA
** Genetic data
@gen: list of 609 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 609 individual labels
@loc.names: 273 locus labels
@loc.all: 273 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 12-516)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, pop, sex, sex_original, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Lichenostomus_HeHo_v1, ChromPos_Lichenostomus_HeHo_v1, AlnCnt_Lichenostomus_HeHo_v1, AlnEvalue_Lichenostomus_HeHo_v1, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, maf
@other$latlon[g]: no coordinates attachedres$autosomal ********************
*** DARTR OBJECT ***
********************
** 609 genotypes, 488 SNPs , size: 49.4 Mb
missing data: 72252 (=24.31 %) scored as NA
** Genetic data
@gen: list of 609 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 609 individual labels
@loc.names: 488 locus labels
@loc.all: 488 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 12-516)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, pop, sex, sex_original, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Lichenostomus_HeHo_v1, ChromPos_Lichenostomus_HeHo_v1, AlnCnt_Lichenostomus_HeHo_v1, AlnEvalue_Lichenostomus_HeHo_v1, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, maf
@other$latlon[g]: no coordinates attachedknitr::kable(head(res$results.table)) # The output table| index | count.F.miss | count.M.miss | count.F.scored | count.M.scored | ratio | p.value | p.adjusted | scoringRate.F | scoringRate.M | w.linked | sex.biased | count.F.het | count.M.het | count.F.hom | count.M.hom | stat | stat.p.value | stat.p.adjusted | heterozygosity.F | heterozygosity.M | z.linked | zw.gametolog | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 27382025-26-T/C | 1 | 61 | 25 | 215 | 308 | 3.4882302 | 0.0000003 | 0.0000016 | 0.7789855 | 0.9249249 | FALSE | TRUE | 0 | 73 | 215 | 235 | NA | NA | NA | 0.0000000 | 0.2370130 | FALSE | FALSE |
| 27338005-34-A/G | 2 | 12 | 13 | 264 | 320 | 1.1186728 | 0.8390268 | 1.0000000 | 0.9565217 | 0.9609610 | FALSE | FALSE | 0 | 144 | 264 | 176 | 0.0046581 | 0.0000000 | 0.0000000 | 0.0000000 | 0.4500000 | TRUE | FALSE |
| 27331627-16-T/G | 3 | 108 | 159 | 168 | 174 | 0.7039235 | 0.0334301 | 0.0860868 | 0.6086957 | 0.5225225 | FALSE | FALSE | 0 | 2 | 168 | 172 | 0.5128690 | 1.0000000 | 1.0000000 | 0.0000000 | 0.0114943 | FALSE | FALSE |
| 53948461-35-G/A | 4 | 46 | 64 | 230 | 269 | 0.8408645 | 0.4593502 | 0.7791707 | 0.8333333 | 0.8078078 | FALSE | FALSE | 29 | 27 | 201 | 242 | 1.2924846 | 0.3950852 | 0.9051780 | 0.1260870 | 0.1003717 | FALSE | FALSE |
| 27360874-8-A/G | 5 | 41 | 63 | 235 | 270 | 0.7480768 | 0.1956613 | 0.4120495 | 0.8514493 | 0.8108108 | FALSE | FALSE | 25 | 38 | 210 | 232 | 0.7272744 | 0.2807852 | 0.7009152 | 0.1063830 | 0.1407407 | FALSE | FALSE |
| 27377678-32-C/A | 6 | 30 | 33 | 246 | 300 | 1.1084581 | 0.7894511 | 1.0000000 | 0.8913043 | 0.9009009 | FALSE | FALSE | 3 | 6 | 243 | 294 | 0.6054740 | 0.5234966 | 1.0000000 | 0.0121951 | 0.0200000 | FALSE | FALSE |
The output consists of a genlight object for each type of loci, plus a results table.
Run gl.infer.sex
This function uses the complete output of function gl.filter.sexlinked (list of 6 objects) to infer the sex of all individuals in the dataset. Specifically, the function uses 3 types of sex-linked loci (W-/Y-linked, Z-/X-linked, and gametologs), assigns a preliminary genetic sex for each type of sex-linked loci available, and outputs an agreed sex.
sexID <- dartR.sexlinked::gl.infer.sex(gl_sex_filtered = res, system = "zw", seed = 124)***FINISHED***knitr::kable(head(sexID))| id | w.linked.sex | #missing | #called | z.linked.sex | #Hom.z | #Het.z | gametolog.sex | #Hom.g | #Het.g | agreed.sex | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ANWC46839 | ANWC46839 | F | 51 | 1 | F | 1 | 141 | F | 5 | 0 | F |
| W49 | W49 | F | 52 | 0 | F | 2 | 156 | F | 5 | 0 | F |
| W90 | W90 | F | 48 | 4 | F | 0 | 162 | F | 5 | 0 | F |
| C25 | C25 | M | 0 | 52 | M | 52 | 113 | M | 0 | 5 | M |
| C8 | C8 | M | 0 | 52 | M | 48 | 116 | M | 0 | 5 | M |
| W70 | W70 | F | 49 | 3 | F | 0 | 152 | F | 5 | 0 | F |
Warning
IMPORTANT We created this function with the explicit intent that a human checks the evidence for the agreed sex that do NOT agree for all types of sex-linked loci (denoted as ‘*M’ or ‘*F’). This human can then use their criterion to validate these assignments.
Exercise
![]()
Can you find individuals for which the agreed sex is uncertain (i.e., has an asterisk “*”)?
Dataset 2 - XX/XY - The Leadbeater’s possum

Load data
#data("LBP")
load('./data/LBP.rda')
LBP # Explore the dataset ********************
*** DARTR OBJECT ***
********************
** 376 genotypes, 1,000 SNPs , size: 5.2 Mb
missing data: 20670 (=5.5 %) scored as NA
** Genetic data
@gen: list of 376 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 376 individual labels
@loc.names: 1000 locus labels
@loc.all: 1000 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 95-281)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, sex, pop, Year.collected, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Possum_v2, ChromPos_Possum_v2, AlnCnt_Possum_v2, AlnEvalue_Possum_v2, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, monomorphs, maf, OneRatio, PIC
@other$latlon[g]: no coordinates attachedLBP@n.loc # Number of SNPs[1] 1000length(LBP@ind.names) # Number of individuals[1] 376Run gl.filter.sexlinked
This function identifies sex-linked and autosomal loci present in a SNP dataset (genlight object) using individuals with known sex. It identifies five types of loci: w-linked or y-linked, sex-biased, z-linked or x-linked, gametologous and autosomal.
The genlight object must contain in gl@other$ind.metrics a column named “id”, and a column named “sex” in which individuals with known-sex are assigned ‘M’ for male, or ‘F’ for female. The function ignores individuals that are assigned anything else or nothing at all (unknown-sex).

knitr::kable(head(LBP@other$ind.metrics)) # Check that ind.metrics has the necessary columns| id | sex | pop | Year.collected | service | plate_location | |
|---|---|---|---|---|---|---|
| Y2 | Y2 | F | Yellingbo | 1997 | DLpos17-2786 | 1-A1 |
| Y16 | Y16 | M | Yellingbo | 2001 | DLpos17-2786 | 1-A10 |
| Y17 | Y17 | F | Yellingbo | 1997 | DLpos17-2786 | 1-A11 |
| Y18 | Y18 | F | Yellingbo | 1999 | DLpos17-2786 | 1-A12 |
| Y3 | Y3 | F | Yellingbo | 1997 | DLpos17-2786 | 1-A2 |
| Y4 | Y4 | M | Yellingbo | 1997 | DLpos17-2786 | 1-A3 |
res <- dartR.sexlinked::gl.filter.sexlinked(gl = LBP, system = "xy")Detected 162 females and 211 males.Starting phase 1. May take a while...Building call rate plots.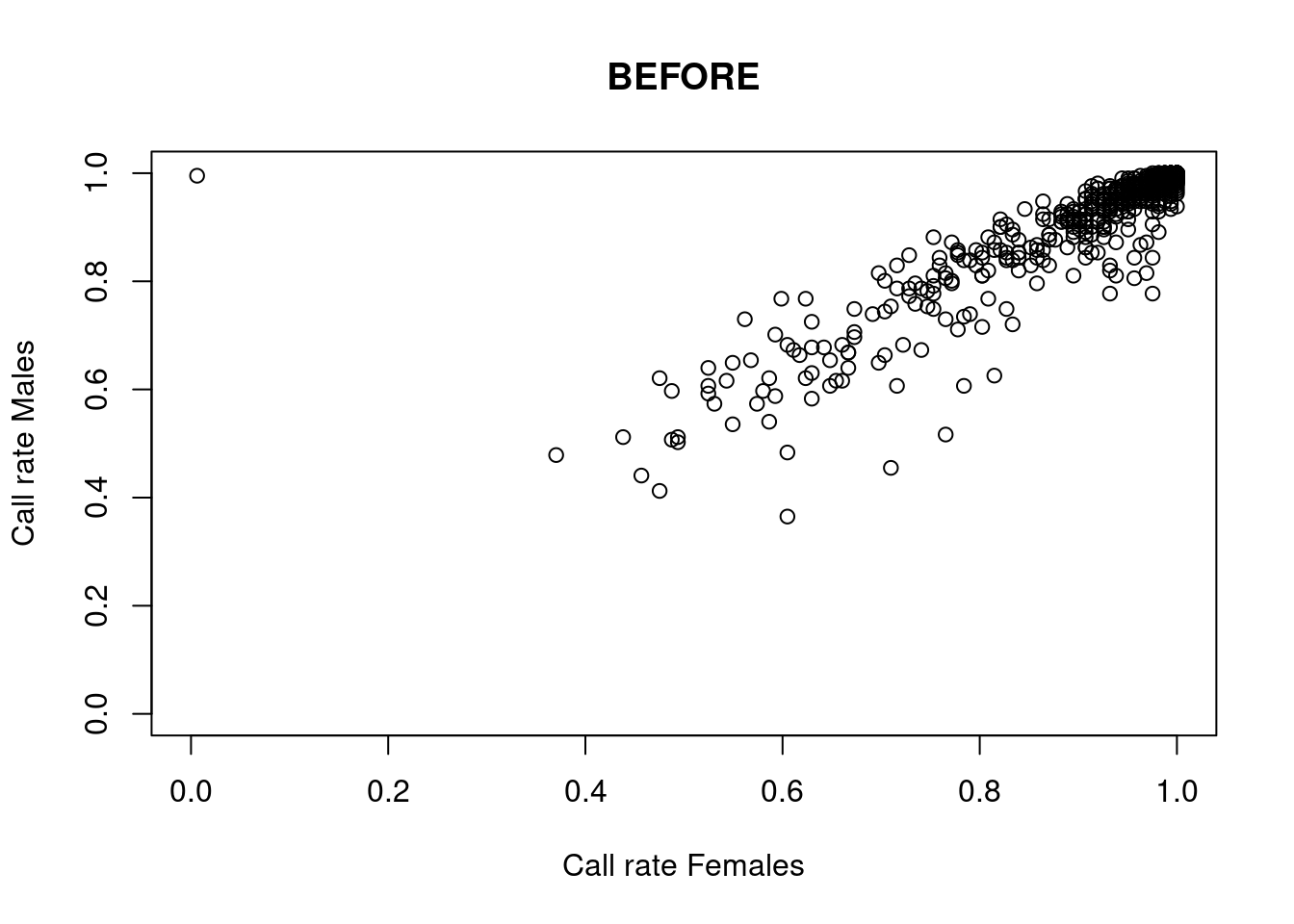
Done. Starting phase 2.Building heterozygosity plots.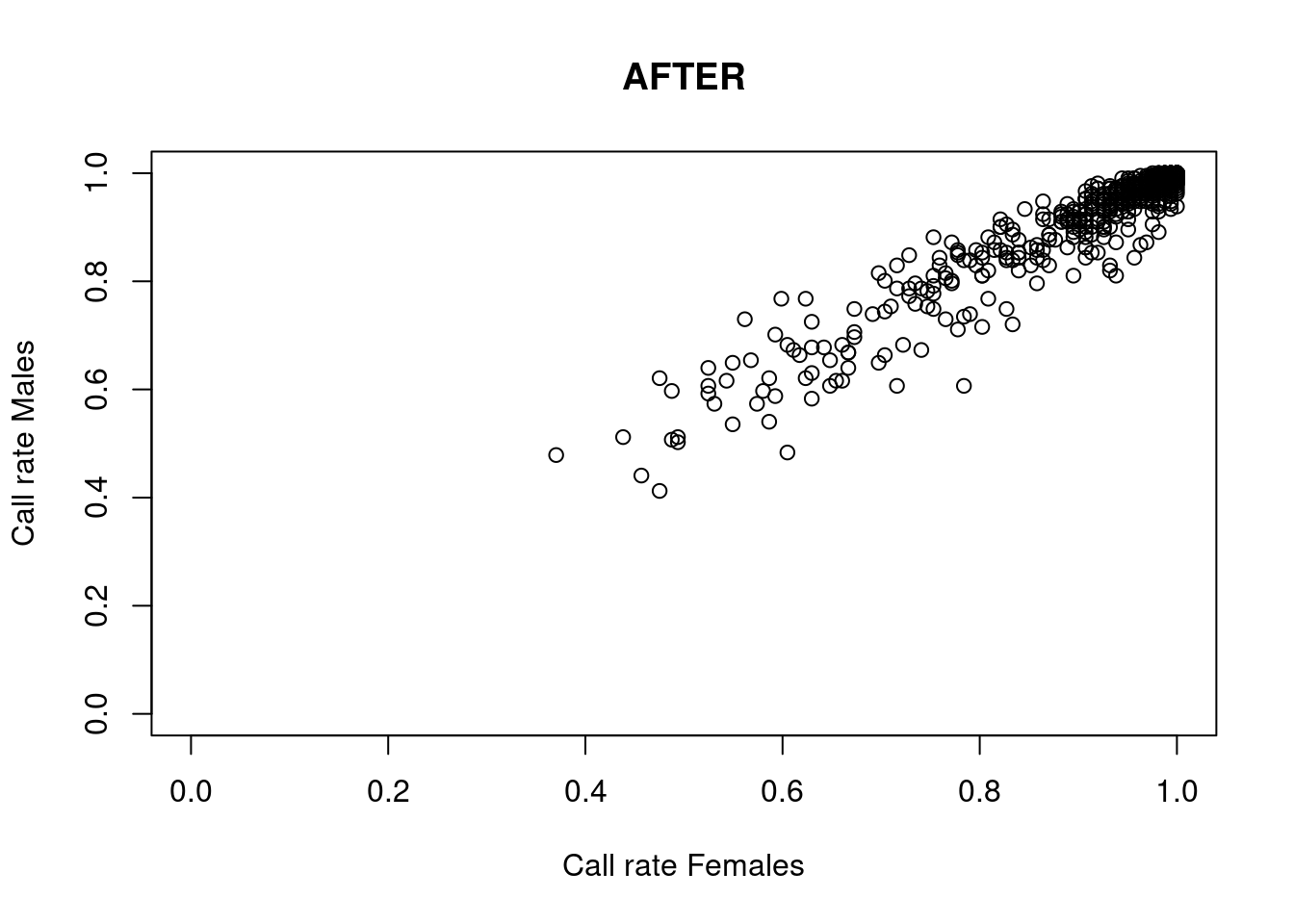
Done building heterozygosity plots.**FINISHED** Total of analyzed loci: 1000.
Found 77 sex-linked loci:
1 Y-linked loci
9 sex-biased loci
66 X-linked loci
1 XY gametologs.
And 923 autosomal loci.
Exercise
![]()
How many males and females does the dataset contain?
How many sex-linked loci were found?
Now check the output:
res$y.linked # Notice that it says "y-linked" ********************
*** DARTR OBJECT ***
********************
** 376 genotypes, 1 SNPs , size: 4.7 Mb
missing data: 164 (=43.62 %) scored as NA
** Genetic data
@gen: list of 376 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 376 individual labels
@loc.names: 1 locus labels
@loc.all: 1 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 95-281)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, sex, pop, Year.collected, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Possum_v2, ChromPos_Possum_v2, AlnCnt_Possum_v2, AlnEvalue_Possum_v2, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, monomorphs, maf, OneRatio, PIC
@other$latlon[g]: no coordinates attachedres$x.linked # Notice that it says "x-linked" ********************
*** DARTR OBJECT ***
********************
** 376 genotypes, 66 SNPs , size: 4.7 Mb
missing data: 827 (=3.33 %) scored as NA
** Genetic data
@gen: list of 376 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 376 individual labels
@loc.names: 66 locus labels
@loc.all: 66 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 95-281)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, sex, pop, Year.collected, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Possum_v2, ChromPos_Possum_v2, AlnCnt_Possum_v2, AlnEvalue_Possum_v2, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, monomorphs, maf, OneRatio, PIC
@other$latlon[g]: no coordinates attachedres$gametolog ********************
*** DARTR OBJECT ***
********************
** 376 genotypes, 1 SNPs , size: 4.7 Mb
missing data: 0 (=0 %) scored as NA
** Genetic data
@gen: list of 376 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 376 individual labels
@loc.names: 1 locus labels
@loc.all: 1 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 95-281)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, sex, pop, Year.collected, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Possum_v2, ChromPos_Possum_v2, AlnCnt_Possum_v2, AlnEvalue_Possum_v2, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, monomorphs, maf, OneRatio, PIC
@other$latlon[g]: no coordinates attachedres$sex.biased ********************
*** DARTR OBJECT ***
********************
** 376 genotypes, 9 SNPs , size: 4.7 Mb
missing data: 853 (=25.21 %) scored as NA
** Genetic data
@gen: list of 376 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 376 individual labels
@loc.names: 9 locus labels
@loc.all: 9 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 95-281)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, sex, pop, Year.collected, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Possum_v2, ChromPos_Possum_v2, AlnCnt_Possum_v2, AlnEvalue_Possum_v2, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, monomorphs, maf, OneRatio, PIC
@other$latlon[g]: no coordinates attachedres$autosomal ********************
*** DARTR OBJECT ***
********************
** 376 genotypes, 923 SNPs , size: 5.2 Mb
missing data: 18826 (=5.42 %) scored as NA
** Genetic data
@gen: list of 376 SNPbin
@ploidy: ploidy of each individual (range: 2-2)
** Additional data
@ind.names: 376 individual labels
@loc.names: 923 locus labels
@loc.all: 923 allele labels
@position: integer storing positions of the SNPs [within 69 base sequence]
@pop: population of each individual (group size range: 95-281)
@other: a list containing: loc.metrics, ind.metrics, loc.metrics.flags, verbose, history
@other$ind.metrics: id, sex, pop, Year.collected, service, plate_location
@other$loc.metrics: AlleleID, CloneID, AlleleSequence, TrimmedSequence, Chrom_Possum_v2, ChromPos_Possum_v2, AlnCnt_Possum_v2, AlnEvalue_Possum_v2, SNP, SnpPosition, CallRate, OneRatioRef, OneRatioSnp, FreqHomRef, FreqHomSnp, FreqHets, PICRef, PICSnp, AvgPIC, AvgCountRef, AvgCountSnp, RepAvg, clone, uid, rdepth, monomorphs, maf, OneRatio, PIC
@other$latlon[g]: no coordinates attachedknitr::kable(head(res$results.table)) # The output table| index | count.F.miss | count.M.miss | count.F.scored | count.M.scored | ratio | p.value | p.adjusted | scoringRate.F | scoringRate.M | y.linked | sex.biased | count.F.het | count.M.het | count.F.hom | count.M.hom | stat | stat.p.value | stat.p.adjusted | heterozygosity.F | heterozygosity.M | x.linked | xy.gametolog | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 28681424-34-G/T | 1 | 0 | 1 | 162 | 210 | 1.2953770 | 1.0000000 | 1 | 1.0000000 | 0.9952607 | FALSE | FALSE | 1 | 0 | 161 | 210 | 1.303397 | 1.0000000 | 1.0000000 | 0.0061728 | 0.0000000 | FALSE | FALSE |
| 28678947-56-C/T | 2 | 12 | 8 | 150 | 203 | 2.0261283 | 0.1638428 | 1 | 0.9259259 | 0.9620853 | FALSE | FALSE | 9 | 7 | 141 | 196 | 1.784196 | 0.3044519 | 0.9598961 | 0.0600000 | 0.0344828 | FALSE | FALSE |
| 28680567-32-T/G | 3 | 12 | 12 | 150 | 199 | 1.3256351 | 0.5289429 | 1 | 0.9259259 | 0.9431280 | FALSE | FALSE | 9 | 11 | 141 | 188 | 1.090635 | 1.0000000 | 1.0000000 | 0.0600000 | 0.0552764 | FALSE | FALSE |
| 28688313-7-C/G | 4 | 0 | 0 | 162 | 211 | 1.3015303 | 1.0000000 | 1 | 1.0000000 | 1.0000000 | FALSE | FALSE | 6 | 0 | 156 | 211 | 8.076192 | 0.0459068 | 0.3917911 | 0.0370370 | 0.0000000 | FALSE | FALSE |
| 28681679-51-C/T | 5 | 22 | 30 | 140 | 181 | 0.9482168 | 0.8814171 | 1 | 0.8641975 | 0.8578199 | FALSE | FALSE | 1 | 1 | 139 | 180 | 1.293900 | 1.0000000 | 1.0000000 | 0.0071429 | 0.0055249 | FALSE | FALSE |
| 28681994-14-G/A | 6 | 0 | 1 | 162 | 210 | 1.2953770 | 1.0000000 | 1 | 1.0000000 | 0.9952607 | FALSE | FALSE | 18 | 19 | 144 | 191 | 1.255791 | 0.6007790 | 1.0000000 | 0.1111111 | 0.0904762 | FALSE | FALSE |
The output consists of a genlight object for each type of loci, plus a results table.
Run gl.infer.sex
This function uses the output of function gl.filter.sexlinked (list of 6 objects) to infer the sex of all individuals in the dataset. It uses 3 types of sex-linked loci (W-/Y-linked, Z-/X-linked, and gametologs), assigns a preliminary genetic sex for each type of sex-linked loci available, and outputs an agreed sex.
sexID <- dartR.sexlinked::gl.infer.sex(gl_sex_filtered = res, system = "xy", seed = 124)Not enough gametologs (need at least 5). Assigning NA...***FINISHED***knitr::kable(head(sexID))| id | y.linked.sex | #called | #missing | x.linked.sex | #Het.x | #Hom.x | gametolog.sex | #Het.g | #Hom.g | agreed.sex | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Y2 | Y2 | F | 0 | 1 | F | 19 | 47 | NA | NA | NA | F |
| Y16 | Y16 | M | 1 | 0 | M | 2 | 56 | NA | NA | NA | M |
| Y17 | Y17 | F | 0 | 1 | F | 27 | 37 | NA | NA | NA | F |
| Y18 | Y18 | F | 0 | 1 | M | 4 | 62 | NA | NA | NA | *F |
| Y3 | Y3 | M | 1 | 0 | M | 3 | 63 | NA | NA | NA | M |
| Y4 | Y4 | M | 1 | 0 | M | 1 | 63 | NA | NA | NA | M |
What did the function mention about gametologs? How did that impact the results table? How many types of sex-linked loci were used to infer an agreed sex?
Exercise
![]()
Can you find individuals for which the agreed sex is uncertain (i.e., has an asterisk “*”)?
EXERCISE
Exercise
![]()
Imagine you are analyzing the genetic data of a population and you want to look at its genetic structure and genetic diversity. You get to work by filtering your beautiful set of SNPs. Because you are a rigorous, you want to test whether using function gl.filter.sexlinked to properly filter out sex-linked loci changes the results of the populations analyses. So you: (1) process your SNP dataset by applying standard filters and run analyses, and (2) process the dataset by filtering out sex-linked loci first, applying standard filters second, and then run analyses.
Choose one of the following datasets (or your own data) and report:
How many sex-linked markers are present?
How many individuals had a wrong sexID?
Do you see any changes in your PCA/structure analyses when you filtered out sex-linked markers versus when you did not?
Do you see any differences in genetic diversity and fixation indices when you filtered out sex-linked markers versus when you did not?




Further study
Exercise data 1 - Your own data
HINT
You can have a look at the exercise data below for inspiration.
Exercise - Your own data
Number of sex-linked markers?
Individuals with wrong sexID?
Changes in PCA before and after removing the SLM?
Differences in genetic diversity and fixation indices between autosomal and SLM?
Exercise data 2 - The Eastern Yellow Robin
Data from Robledo-Ruiz et al. (2023)
Load data
#data("EYR")
load('./data/EYR.rda')
EYR@n.loc
table(EYR@pop)
table(EYR@other$ind.metrics$pop)
table(EYR@other$ind.metrics$sex, useNA = "ifany")[1] 1000
Crusoe Muckleford Timor Wombat
238 421 52 71
Crusoe Muckleford Timor Wombat
238 421 52 71
F M
1 352 429 1. Number of sex-linked markers?
res <- dartR.sexlinked::gl.filter.sexlinked(gl = EYR, system = "zw")Detected 352 females and 429 males.Starting phase 1. May take a while...Building call rate plots.
Done. Starting phase 2.Building heterozygosity plots.
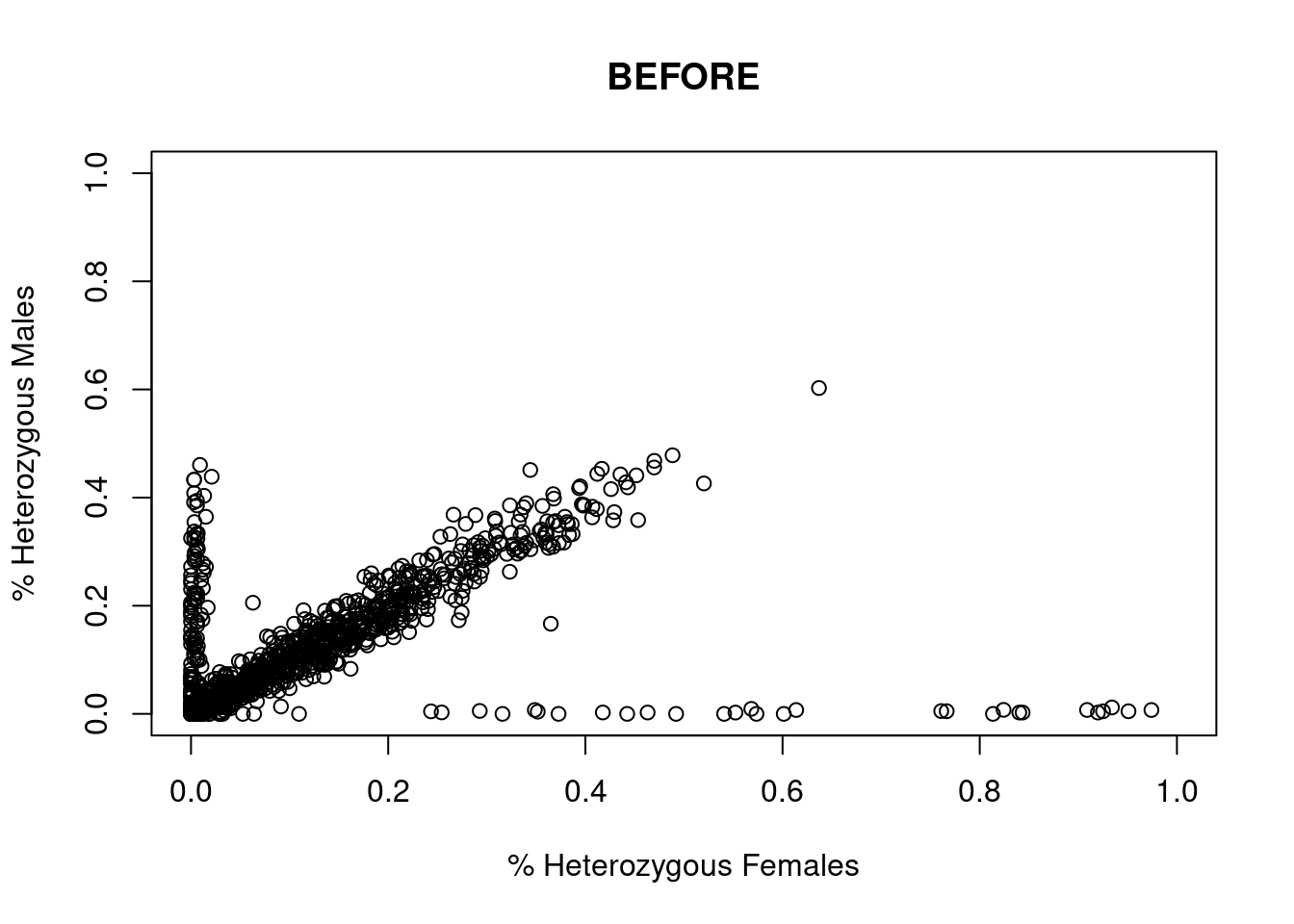
Done building heterozygosity plots.**FINISHED** Total of analyzed loci: 1000.
Found 150 sex-linked loci:
16 W-linked loci
82 sex-biased loci
32 Z-linked loci
20 ZW gametologs.
And 850 autosomal loci.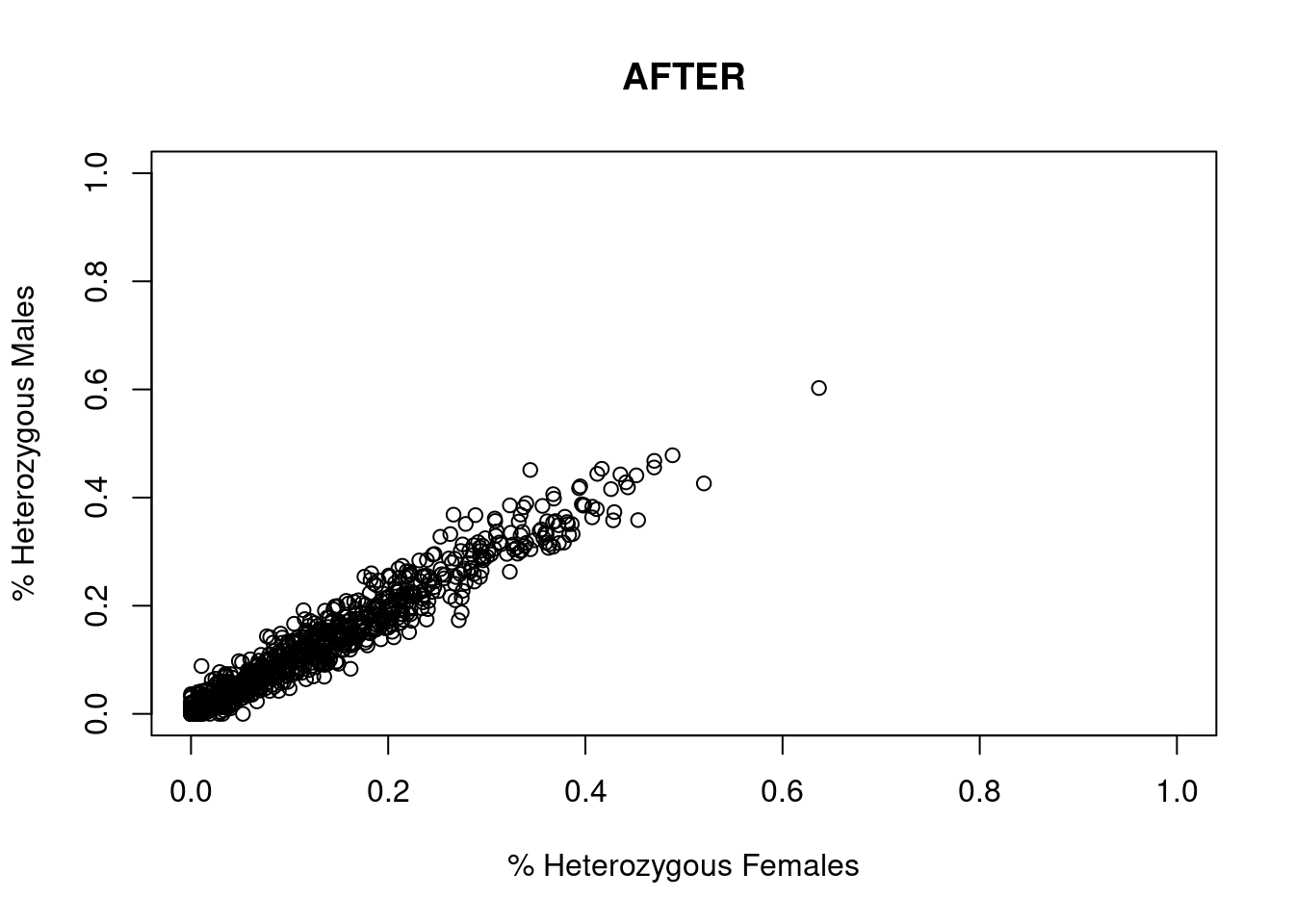
2. Individuals with wrong sexID?
sexID <- dartR.sexlinked::gl.infer.sex(gl_sex_filtered = res, system = "zw", seed = 124)***FINISHED***knitr::kable(head(sexID))
sum(EYR$other$ind.metrics$sex != sexID$agreed.sex, na.rm = TRUE)| id | w.linked.sex | #missing | #called | z.linked.sex | #Hom.z | #Het.z | gametolog.sex | #Hom.g | #Het.g | agreed.sex | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 024-96401 | 024-96401 | M | 0 | 16 | M | 7 | 25 | M | 0 | 5 | M |
| 024-96401b | 024-96401b | M | 0 | 16 | M | 9 | 21 | M | 0 | 5 | M |
| 024-96402 | 024-96402 | F | 15 | 1 | F | 0 | 32 | F | 5 | 0 | F |
| 024-96403 | 024-96403 | M | 1 | 15 | M | 11 | 21 | M | 0 | 5 | M |
| 024-96404 | 024-96404 | M | 0 | 16 | M | 12 | 20 | M | 0 | 5 | M |
| 024-96405 | 024-96405 | M | 0 | 16 | M | 11 | 21 | M | 0 | 5 | M |
[1] 55
Exercise
![]()
Can you tell which misidentified sexes are due to uncertain genetic sex (indicated with *)?
HINT Try using grep(pattern = "\\*", x = sexID$agreed.sex)
Processing SNPs with two filtering regimes
Filtering SNPs only with standard filters (sloppy)
# Filter for read depth
dartR.base::gl.report.rdepth(EYR) # This is the initial datasetStarting ::
Starting dartR.base
Starting gl.report.rdepth
Processing genlight object with SNP data
Reporting Read Depth by Locus
No. of loci = 1000
No. of individuals = 782
Minimum : 2.6
1st quartile : 4.3
Median : 5.6
Mean : 5.9649
3r quartile : 7.325
Maximum : 13.2
Missing Rate Overall: 0.19 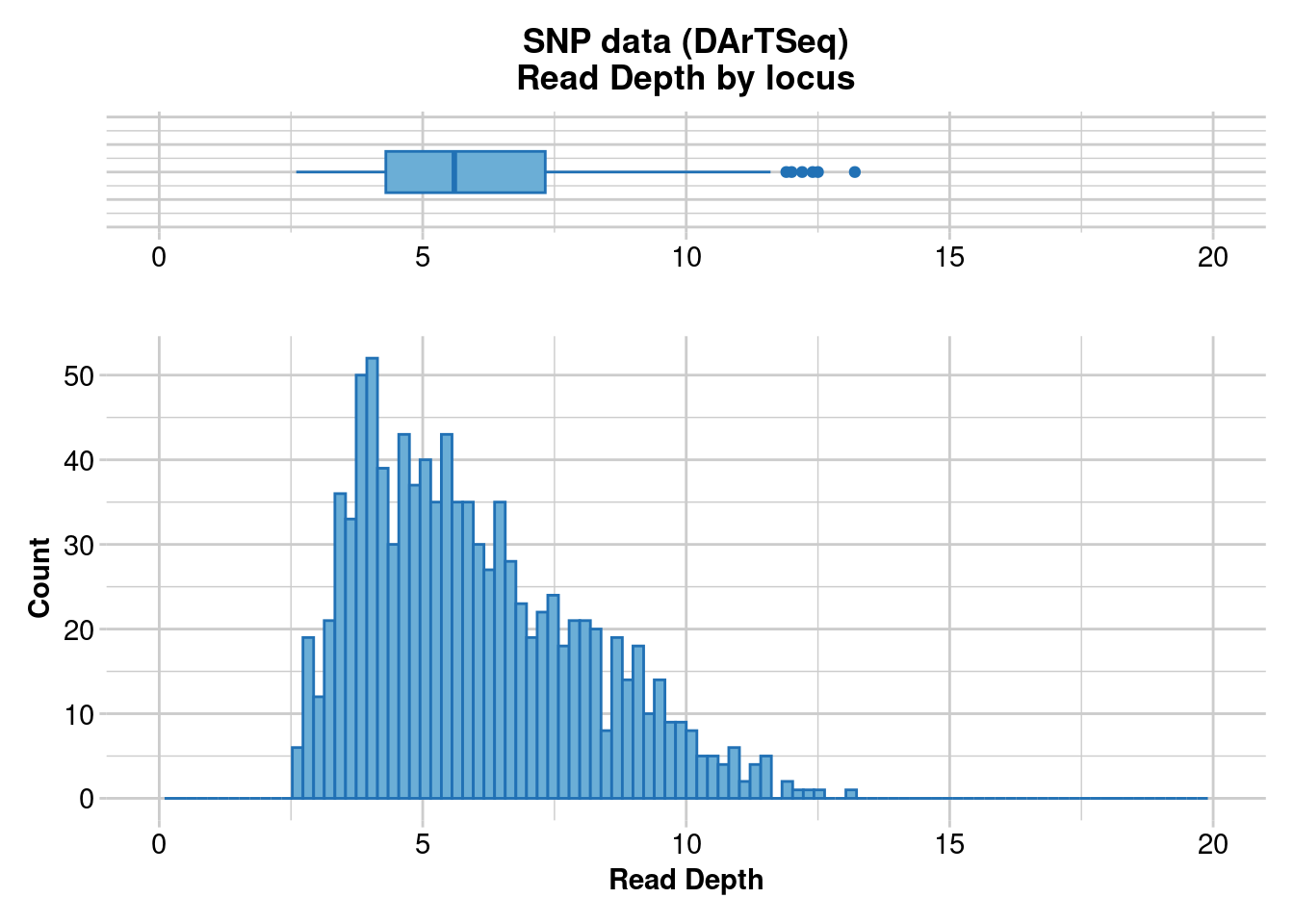
Quantile Threshold Retained Percent Filtered Percent
1 100% 13.2 1 0.1 999 99.9
2 95% 9.9 51 5.1 949 94.9
3 90% 9.0 105 10.5 895 89.5
4 85% 8.3 151 15.1 849 84.9
5 80% 7.8 208 20.8 792 79.2
6 75% 7.3 258 25.8 742 74.2
7 70% 6.9 304 30.4 696 69.6
8 65% 6.5 354 35.4 646 64.6
9 60% 6.2 404 40.4 596 59.6
10 55% 5.9 451 45.1 549 54.9
11 50% 5.6 504 50.4 496 49.6
12 45% 5.3 563 56.3 437 43.7
13 40% 5.1 602 60.2 398 39.8
14 35% 4.8 659 65.9 341 34.1
15 30% 4.6 702 70.2 298 29.8
16 25% 4.3 752 75.2 248 24.8
17 20% 4.0 823 82.3 177 17.7
18 15% 3.9 852 85.2 148 14.8
19 10% 3.6 906 90.6 94 9.4
20 5% 3.3 956 95.6 44 4.4
21 0% 2.6 1000 100.0 0 0.0
Completed: ::
Completed: dartR.base
Completed: gl.report.rdepth EYR.sloppy <- dartR.base::gl.filter.rdepth(EYR, lower = 3, upper = 11, verbose = 0)
# Filter for loci call rate
dartR.base::gl.report.callrate(EYR.sloppy, method = "loc")Starting ::
Starting dartR.base
Starting gl.report.callrate
Processing genlight object with SNP data
Reporting Call Rate by Locus
No. of loci = 958
No. of individuals = 782
Minimum : 0.20844
1st quartile : 0.7202688
Median : 0.895141
Mean : 0.8131871
3r quartile : 0.950128
Maximum : 0.988491
Missing Rate Overall: 0.1868 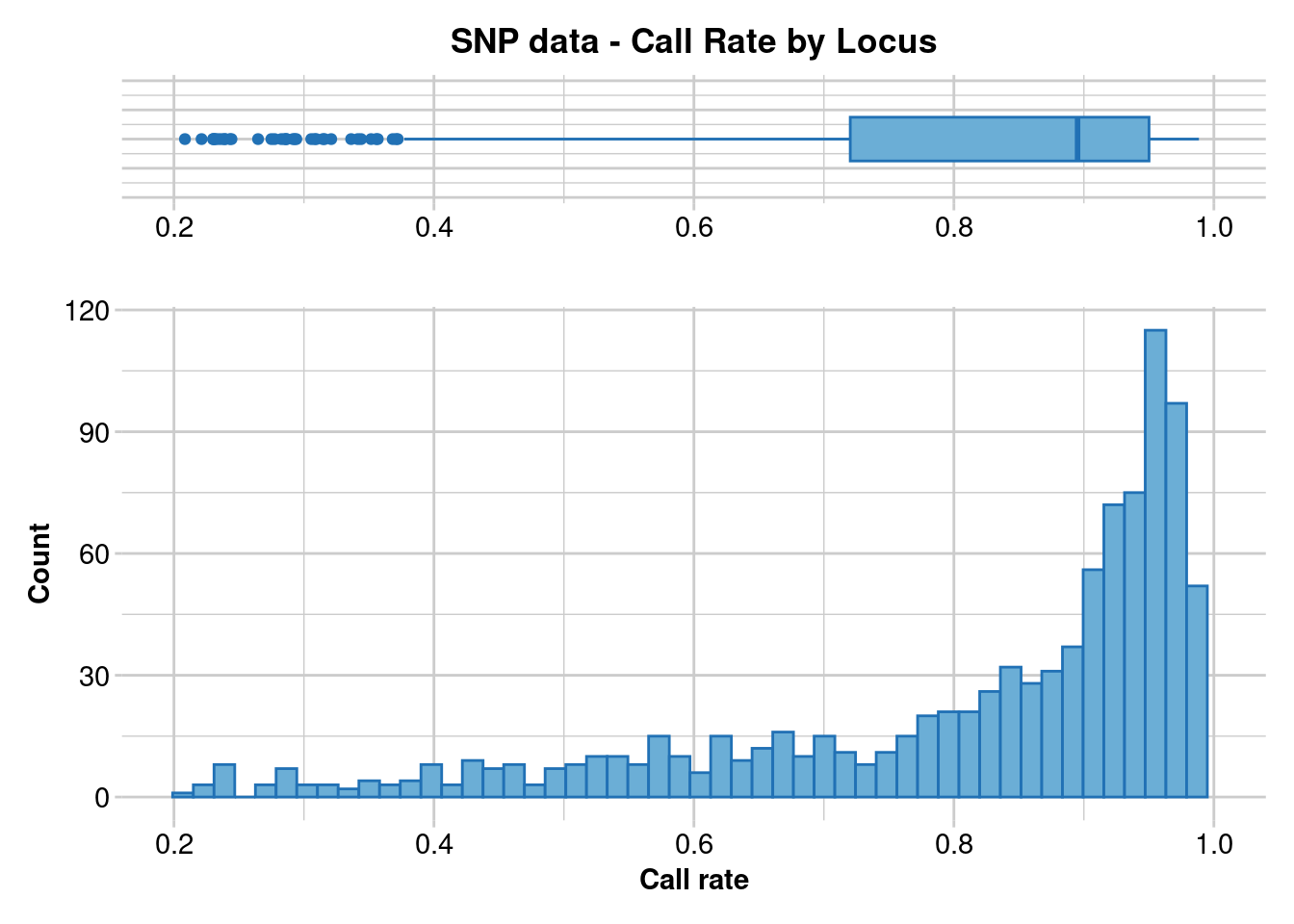
Completed: ::
Completed: dartR.base
Completed: gl.report.callrate EYR.sloppy <- dartR.base::gl.filter.callrate(EYR.sloppy, method = "loc", threshold = 0.75, verbose = 0, recalc = TRUE)
# Filter for individual call rate
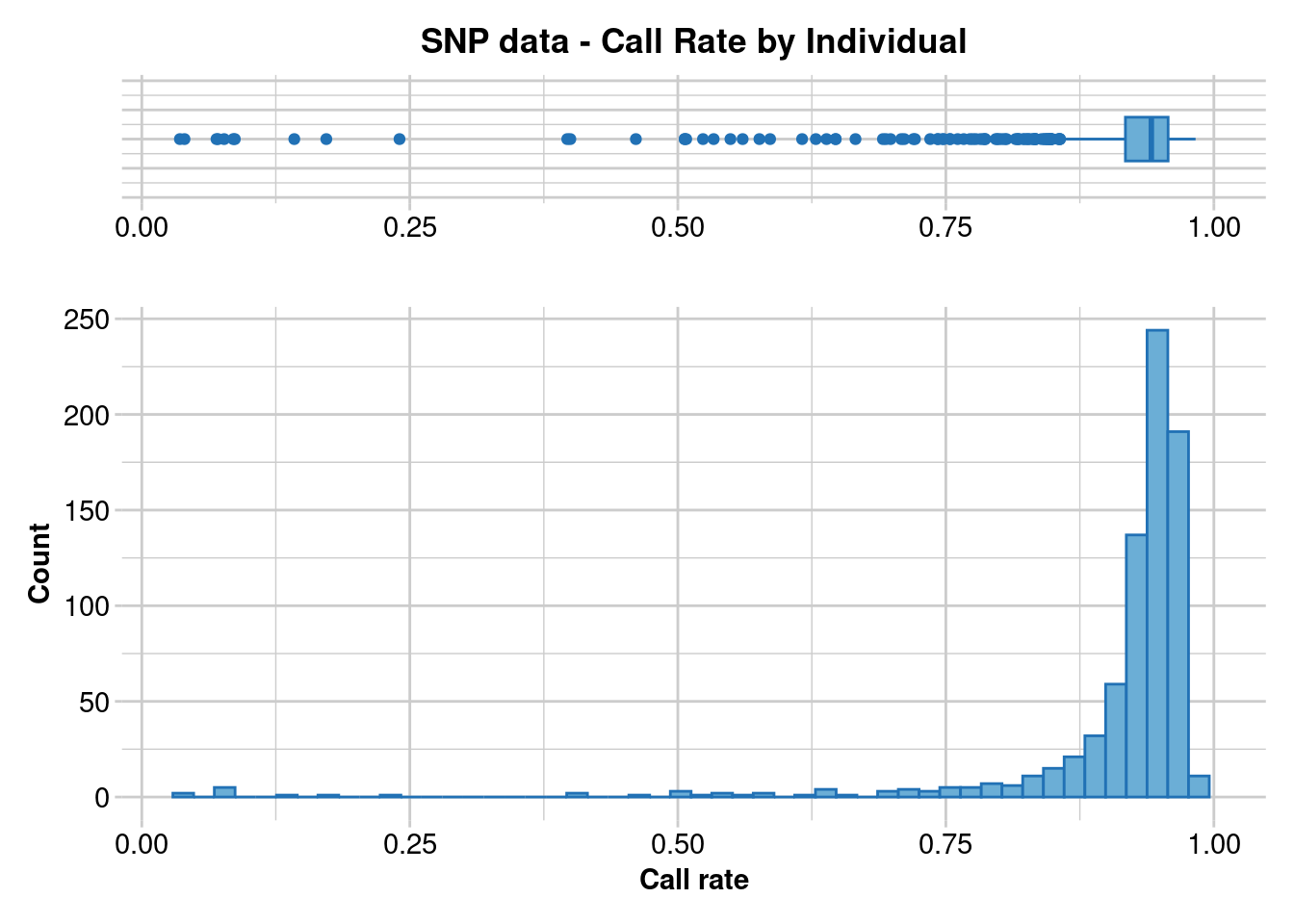
dartR.base::gl.report.callrate(EYR.sloppy, method = "ind")Starting ::
Starting dartR.base
Starting gl.report.callrate
Processing genlight object with SNP data
Reporting Call Rate by Individual
No. of loci = 703
No. of individuals = 782
Minimum : 0.03556188
1st quartile : 0.9174964
Median : 0.9416785
Mean : 0.9108097
3r quartile : 0.9573257
Maximum : 0.9829303
Missing Rate Overall: 0.0892
Listing 4 populations and their average CallRates
Monitor again after filtering
Population CallRate N
1 Crusoe 0.9027 238
2 Muckleford 0.9073 421
3 Timor 0.9402 52
4 Wombat 0.9371 71
Listing 20 individuals with the lowest CallRates
Use this list to see which individuals will be lost on filtering by individual
Set ind.to.list parameter to see more individuals
Individual CallRate
1 M18.29.1 0.03556188
2 M18.18.1 0.03982930
3 M18.47.2 0.06970128
4 C18.16.1 0.07112376
5 027-34168 0.07681366
6 C18.15.2 0.08534851
7 C18.21.2 0.08677098
8 M18.47.3 0.14224751
9 M18.35.2 0.17211949
10 M18.20.3 0.24039829
11 M20.70.2 0.39687055
12 C18.28.1 0.39971550
13 C18.17.2 0.46088193
14 027-34065 0.50640114
15 C18.14.1 0.50640114
16 M20.70.3 0.50782361
17 M20.110.1 0.52347084
18 M19.12.1 0.53342817
19 M19.8.1 0.54907539
20 M20.64.3 0.56045519
)
Completed: ::
Completed: dartR.base
Completed: gl.report.callrate EYR.sloppy <- dartR.base::gl.filter.callrate(EYR.sloppy, method = "ind", threshold = 0.65, verbose = 0, recalc = TRUE)# Filter for MAC (= 3)
dartR.base::gl.report.maf(EYR.sloppy)Starting ::
Starting dartR.base
Starting gl.report.maf
Processing genlight object with SNP data
Starting ::
Starting dartR.base
Starting gl.report.maf
Reporting Minor Allele Frequency (MAF) by Locus for population Crusoe
No. of loci = 670
No. of individuals = 231
Minimum : 0.0022
1st quantile : 0.064825
Median : 0.1582
Mean : 0.1793525
3r quantile : 0.267475
Maximum : 0.4975
Missing Rate Overall: 0.08
Reporting Minor Allele Frequency (MAF) by Locus for population Muckleford
No. of loci = 683
No. of individuals = 401
Minimum : 0.0013
1st quantile : 0.05875
Median : 0.1404
Mean : 0.172949
3r quantile : 0.2617
Maximum : 0.4985
Missing Rate Overall: 0.07
Reporting Minor Allele Frequency (MAF) by Locus for population Timor
No. of loci = 589
No. of individuals = 52
Minimum : 0.0096
1st quantile : 0.0673
Median : 0.1667
Mean : 0.1914129
3r quantile : 0.2872
Maximum : 0.5
Missing Rate Overall: 0.06
Reporting Minor Allele Frequency (MAF) by Locus for population Wombat
No. of loci = 627
No. of individuals = 71
Minimum : 0.007
1st quantile : 0.06385
Median : 0.1449
Mean : 0.1746703
3r quantile : 0.2542
Maximum : 0.5
Missing Rate Overall: 0.06
Reporting Minor Allele Frequency (MAF) by Locus OVERALL
No. of loci = 703
No. of individuals = 755
Minimum : 3e-04
1st quantile : 0.0627
Median : 0.13435
Mean : 0.1696497
3r quantile : 0.246025
Maximum : 0.4991
Missing Rate Overall: 0.07 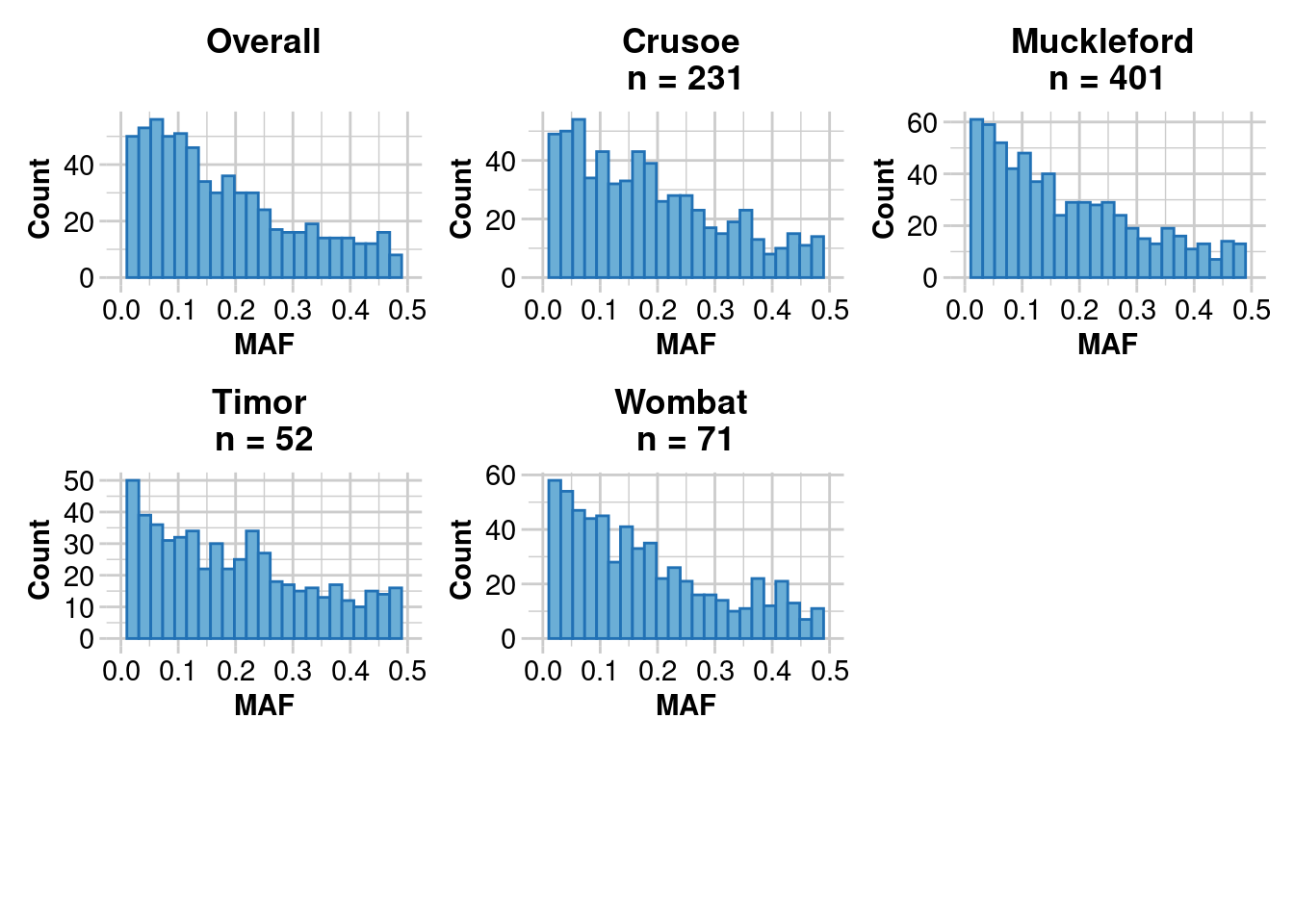
Quantile Threshold Retained Percent Filtered Percent
1 100% 0.4991 1 0.1 699 99.9
2 95% 0.4343 36 5.1 664 94.9
3 90% 0.3807 71 10.1 629 89.9
4 85% 0.3331 105 15.0 595 85.0
5 80% 0.2858 141 20.1 559 79.9
6 75% 0.2460 176 25.1 524 74.9
7 70% 0.2233 210 30.0 490 70.0
8 65% 0.2003 246 35.1 454 64.9
9 60% 0.1797 280 40.0 420 60.0
10 55% 0.1562 315 45.0 385 55.0
11 50% 0.1341 352 50.3 348 49.7
12 45% 0.1214 386 55.1 314 44.9
13 40% 0.1032 421 60.1 279 39.9
14 35% 0.0904 455 65.0 245 35.0
15 30% 0.0742 490 70.0 210 30.0
16 25% 0.0627 526 75.1 174 24.9
17 20% 0.0476 561 80.1 139 19.9
18 15% 0.0359 595 85.0 105 15.0
19 10% 0.0224 631 90.1 69 9.9
20 5% 0.0059 666 95.1 34 4.9
21 0% 0.0003 700 100.0 0 0.0
Completed: ::
Completed: dartR.base
Completed: gl.report.maf EYR.sloppy <- dartR.base::gl.filter.maf(EYR.sloppy, threshold = 3, verbose = 0, recalc = TRUE)Starting gl.select.colors
Warning: Number of required colors not specified, set to 9
Library: RColorBrewer
Palette: brewer.pal
Showing and returning 2 of 9 colors for library RColorBrewer : palette Blues 
Completed: gl.select.colors Filtering SNPs with gl.filter.sexlinked and standard filters (correct)
# Filter for sex-linked loci
correct <- dartR.sexlinked::gl.filter.sexlinked(EYR, system = "zw") # This is the initial dataset
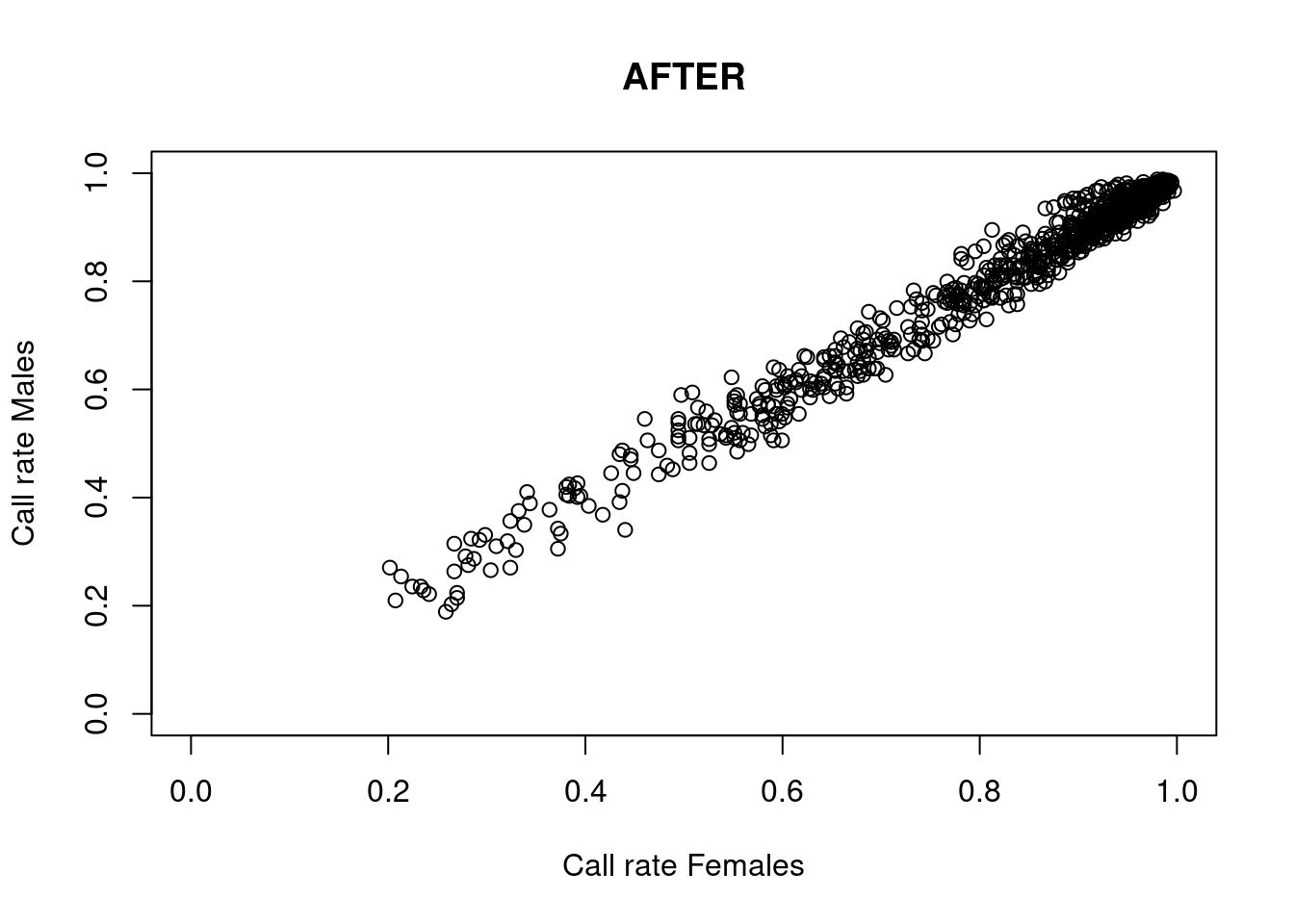


# We will use correct$autosomal for the next filters
# Filter for read depth
dartR.base::gl.report.rdepth(correct$autosomal) # This is the filtered datasetStarting ::
Starting dartR.base
Starting gl.report.rdepth
Processing genlight object with SNP data
Reporting Read Depth by Locus
No. of loci = 850
No. of individuals = 782
Minimum : 2.6
1st quartile : 4.3
Median : 5.6
Mean : 6.008941
3r quartile : 7.4
Maximum : 13.2
Missing Rate Overall: 0.18 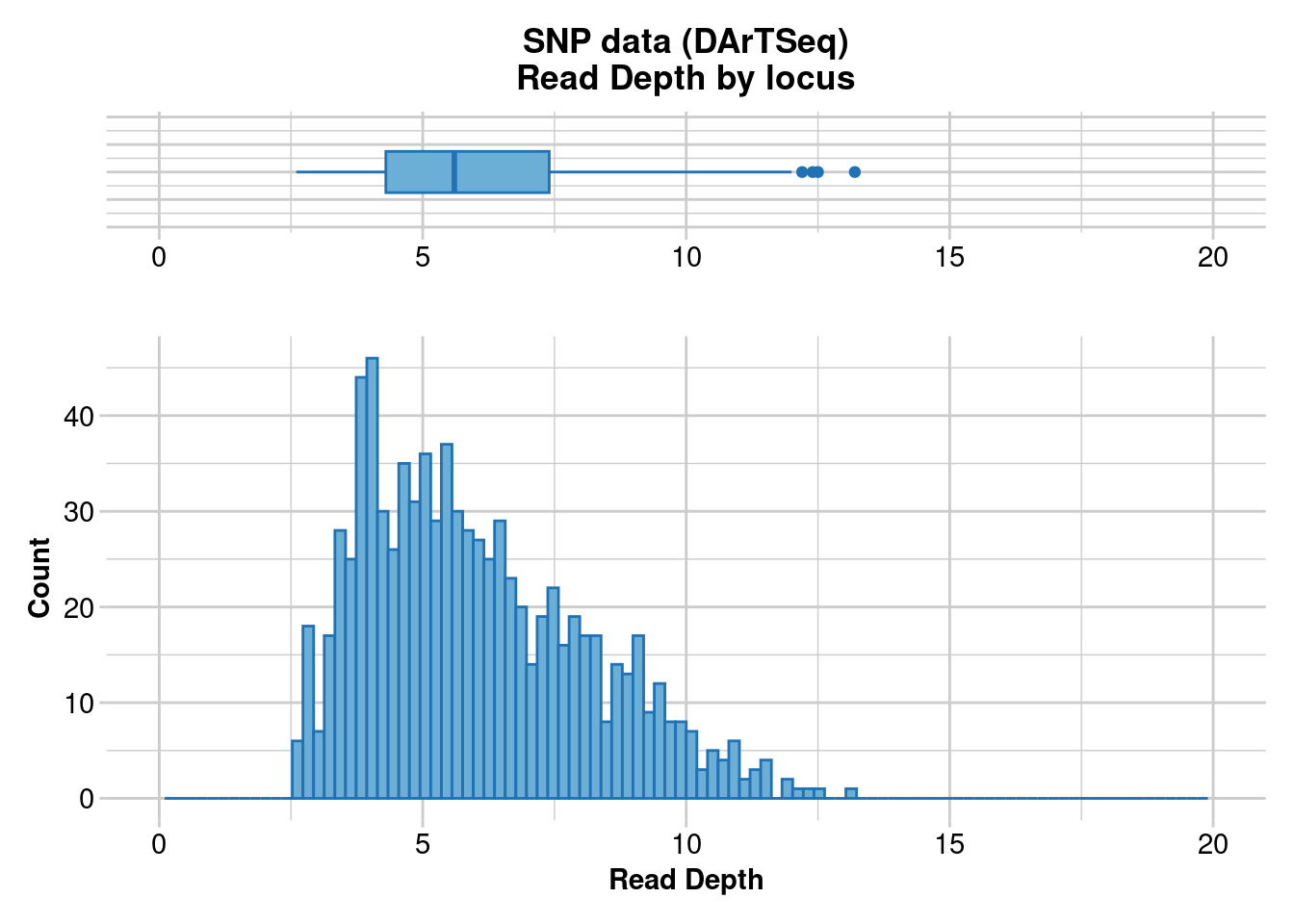
Quantile Threshold Retained Percent Filtered Percent
1 100% 13.2 1 0.1 849 99.9
2 95% 9.9 45 5.3 805 94.7
3 90% 9.1 88 10.4 762 89.6
4 85% 8.4 129 15.2 721 84.8
5 80% 7.9 173 20.4 677 79.6
6 75% 7.4 220 25.9 630 74.1
7 70% 6.9 264 31.1 586 68.9
8 65% 6.5 305 35.9 545 64.1
9 60% 6.2 350 41.2 500 58.8
10 55% 5.9 391 46.0 459 54.0
11 50% 5.6 435 51.2 415 48.8
12 45% 5.4 472 55.5 378 44.5
13 40% 5.1 519 61.1 331 38.9
14 35% 4.9 555 65.3 295 34.7
15 30% 4.6 603 70.9 247 29.1
16 25% 4.3 645 75.9 205 24.1
17 20% 4.0 705 82.9 145 17.1
18 15% 3.9 730 85.9 120 14.1
19 10% 3.6 774 91.1 76 8.9
20 5% 3.3 812 95.5 38 4.5
21 0% 2.6 850 100.0 0 0.0
Completed: ::
Completed: dartR.base
Completed: gl.report.rdepth EYR.correct <- dartR.base::gl.filter.rdepth(correct$autosomal, lower = 3, upper = 11, verbose = 0)
# Filter for loci call rate
dartR.base::gl.report.callrate(EYR.correct, method = "loc")Starting ::
Starting dartR.base
Starting gl.report.callrate
Processing genlight object with SNP data
Reporting Call Rate by Locus
No. of loci = 811
No. of individuals = 782
Minimum : 0.20844
1st quartile : 0.7436065
Median : 0.900256
Mean : 0.8192658
3r quartile : 0.951407
Maximum : 0.988491
Missing Rate Overall: 0.1807 
Completed: ::
Completed: dartR.base
Completed: gl.report.callrate EYR.correct <- dartR.base::gl.filter.callrate(EYR.correct, method = "loc", threshold = 0.75, verbose = 0, recalc = TRUE)
# Filter for individual call rate
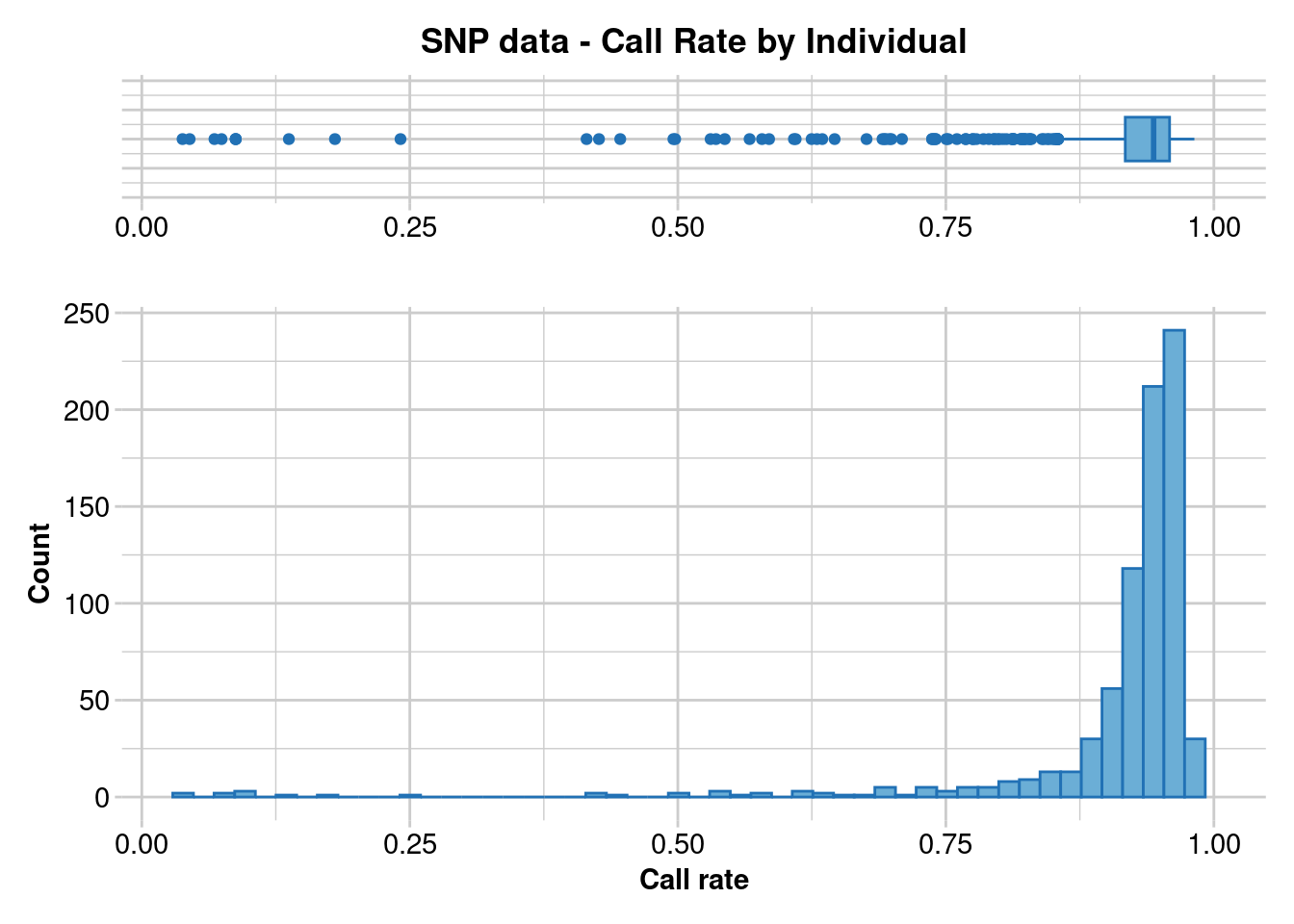
dartR.base::gl.report.callrate(EYR.correct, method = "ind")Starting ::
Starting dartR.base
Starting gl.report.callrate
Processing genlight object with SNP data
Reporting Call Rate by Individual
No. of loci = 605
No. of individuals = 782
Minimum : 0.03801653
1st quartile : 0.9173554
Median : 0.9438017
Mean : 0.9120479
3r quartile : 0.9586777
Maximum : 0.9818182
Missing Rate Overall: 0.088
Listing 4 populations and their average CallRates
Monitor again after filtering
Population CallRate N
1 Crusoe 0.9037 238
2 Muckleford 0.9090 421
3 Timor 0.9418 52
4 Wombat 0.9365 71
Listing 20 individuals with the lowest CallRates
Use this list to see which individuals will be lost on filtering by individual
Set ind.to.list parameter to see more individuals
Individual CallRate
1 M18.29.1 0.03801653
2 M18.18.1 0.04462810
3 M18.47.2 0.06776860
4 C18.16.1 0.07438017
5 027-34168 0.08760331
6 C18.15.2 0.08760331
7 C18.21.2 0.08760331
8 M18.47.3 0.13719008
9 M18.35.2 0.18016529
10 M18.20.3 0.24132231
11 C18.28.1 0.41487603
12 M20.70.2 0.42644628
13 C18.17.2 0.44628099
14 027-34065 0.49586777
15 C18.14.1 0.49752066
16 M20.110.1 0.53057851
17 M20.70.3 0.53553719
18 M19.12.1 0.54380165
19 M19.8.1 0.56694215
20 M19.33.2 0.57851240
)
Completed: ::
Completed: dartR.base
Completed: gl.report.callrate EYR.correct <- dartR.base::gl.filter.callrate(EYR.correct, method = "ind", threshold = 0.65, verbose = 0, recalc = TRUE)# Filter for MAC (= 3)
dartR.base::gl.report.maf(EYR.correct)Starting ::
Starting dartR.base
Starting gl.report.maf
Processing genlight object with SNP data
Starting ::
Starting dartR.base
Starting gl.report.maf
Reporting Minor Allele Frequency (MAF) by Locus for population Crusoe
No. of loci = 573
No. of individuals = 231
Minimum : 0.0022
1st quantile : 0.06
Median : 0.1488
Mean : 0.1741178
3r quantile : 0.2646
Maximum : 0.4975
Missing Rate Overall: 0.08
Reporting Minor Allele Frequency (MAF) by Locus for population Muckleford
No. of loci = 585
No. of individuals = 401
Minimum : 0.0013
1st quantile : 0.055
Median : 0.129
Mean : 0.163993
3r quantile : 0.2474
Maximum : 0.4985
Missing Rate Overall: 0.07
Reporting Minor Allele Frequency (MAF) by Locus for population Timor
No. of loci = 504
No. of individuals = 52
Minimum : 0.0096
1st quantile : 0.068275
Median : 0.1635
Mean : 0.1898613
3r quantile : 0.286075
Maximum : 0.5
Missing Rate Overall: 0.06
Reporting Minor Allele Frequency (MAF) by Locus for population Wombat
No. of loci = 536
No. of individuals = 71
Minimum : 0.007
1st quantile : 0.062475
Median : 0.13805
Mean : 0.1706063
3r quantile : 0.2509
Maximum : 0.5
Missing Rate Overall: 0.06
Reporting Minor Allele Frequency (MAF) by Locus OVERALL
No. of loci = 605
No. of individuals = 755
Minimum : 3e-04
1st quantile : 0.058025
Median : 0.1306
Mean : 0.1628656
3r quantile : 0.23785
Maximum : 0.4991
Missing Rate Overall: 0.07 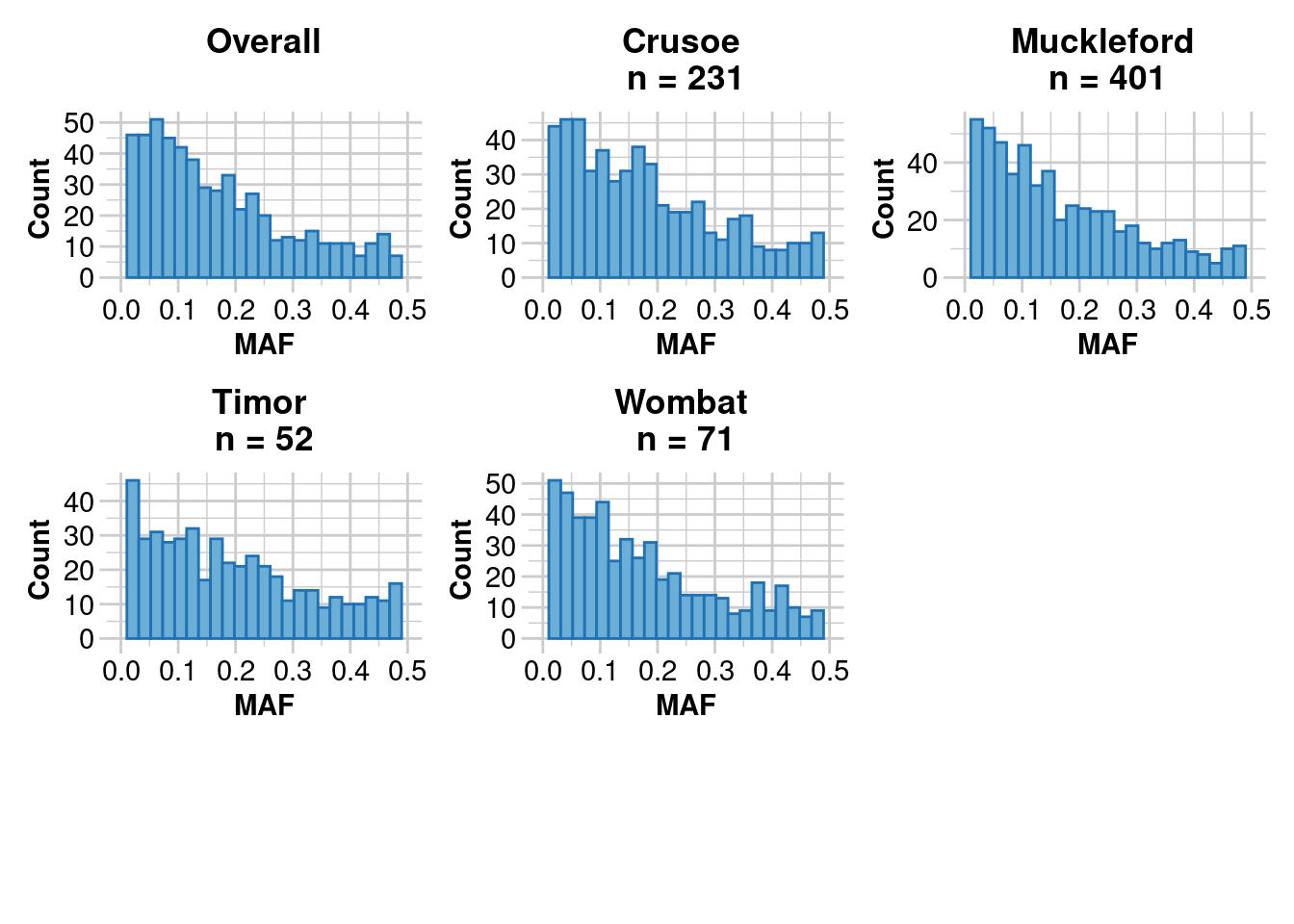
Quantile Threshold Retained Percent Filtered Percent
1 100% 0.4991 1 0.2 601 99.8
2 95% 0.4367 31 5.1 571 94.9
3 90% 0.3771 61 10.1 541 89.9
4 85% 0.3250 91 15.1 511 84.9
5 80% 0.2747 121 20.1 481 79.9
6 75% 0.2387 151 25.1 451 74.9
7 70% 0.2132 181 30.1 421 69.9
8 65% 0.1916 211 35.0 391 65.0
9 60% 0.1720 241 40.0 361 60.0
10 55% 0.1486 271 45.0 331 55.0
11 50% 0.1306 302 50.2 300 49.8
12 45% 0.1118 332 55.1 270 44.9
13 40% 0.0975 362 60.1 240 39.9
14 35% 0.0808 392 65.1 210 34.9
15 30% 0.0700 422 70.1 180 29.9
16 25% 0.0578 452 75.1 150 24.9
17 20% 0.0445 482 80.1 120 19.9
18 15% 0.0300 512 85.0 90 15.0
19 10% 0.0174 542 90.0 60 10.0
20 5% 0.0046 572 95.0 30 5.0
21 0% 0.0003 602 100.0 0 0.0
Completed: ::
Completed: dartR.base
Completed: gl.report.maf EYR.correct <- dartR.base::gl.filter.maf(EYR.correct, threshold = 3, verbose = 0, recalc = TRUE)Starting gl.select.colors
Warning: Number of required colors not specified, set to 9
Library: RColorBrewer
Palette: brewer.pal
Showing and returning 2 of 9 colors for library RColorBrewer : palette Blues 
Completed: gl.select.colors 3. Changes in PCA before and after removing the SLM?
PCA on sloppy dataset (only standard filters)
PCA.sloppy <- dartR.base::gl.pcoa(EYR.sloppy, verbose = 0)
dartR.base::gl.pcoa.plot(PCA.sloppy, EYR.sloppy, xaxis = 1, yaxis = 2)
dartR.base::gl.pcoa.plot(PCA.sloppy, EYR.sloppy, xaxis = 2, yaxis = 3)Starting gl.colors
Selected color type 2
Completed: gl.colors
Starting ::
Starting dartR.base
Starting gl.pcoa.plot
Processing an ordination file (glPca)
Processing genlight object with SNP data
Package directlabels needed for this function to work. Please install it.
[1] -1
Starting ::
Starting dartR.base
Starting gl.pcoa.plot
Processing an ordination file (glPca)
Processing genlight object with SNP data
Package directlabels needed for this function to work. Please install it.
[1] -1PCA on correct dataset (gl.filter.sexlinked and standard filters)
PCA.correct <- dartR.base::gl.pcoa(EYR.correct, verbose = 0)
dartR.base::gl.pcoa.plot(PCA.correct, EYR.correct, xaxis = 1, yaxis = 2)
dartR.base::gl.pcoa.plot(PCA.correct, EYR.correct, xaxis = 2, yaxis = 3)Starting gl.colors
Selected color type 2
Completed: gl.colors
Starting ::
Starting dartR.base
Starting gl.pcoa.plot
Processing an ordination file (glPca)
Processing genlight object with SNP data
Package directlabels needed for this function to work. Please install it.
[1] -1
Starting ::
Starting dartR.base
Starting gl.pcoa.plot
Processing an ordination file (glPca)
Processing genlight object with SNP data
Package directlabels needed for this function to work. Please install it.
[1] -14. Differences in genetic diversity and fixation indices between autosomal and SLM?
# Basic stats
basic.sloppy <- dartR.base::utils.basic.stats(EYR.sloppy)
basic.correct <- dartR.base::utils.basic.stats(EYR.correct)
basic.sloppy$overall Ho Hs Ht Dst Htp Dstp Fst Fstp Fis Dest
0.1603 0.2376 0.2464 0.0087 0.2487 0.0148 0.0355 0.0596 0.3256 0.0259
Gst_max Gst_H
0.7141 0.0834 basic.correct$overall Ho Hs Ht Dst Htp Dstp Fst Fstp Fis Dest
0.1579 0.2302 0.2375 0.0073 0.2398 0.0128 0.0307 0.0532 0.3139 0.0221
Gst_max Gst_H
0.7229 0.0737 # Genetic diversity per pop
divers.sloppy <- dartR.base::gl.report.diversity(EYR.sloppy, pbar = FALSE, table = FALSE, verbose = 0) Processing genlight object with SNP data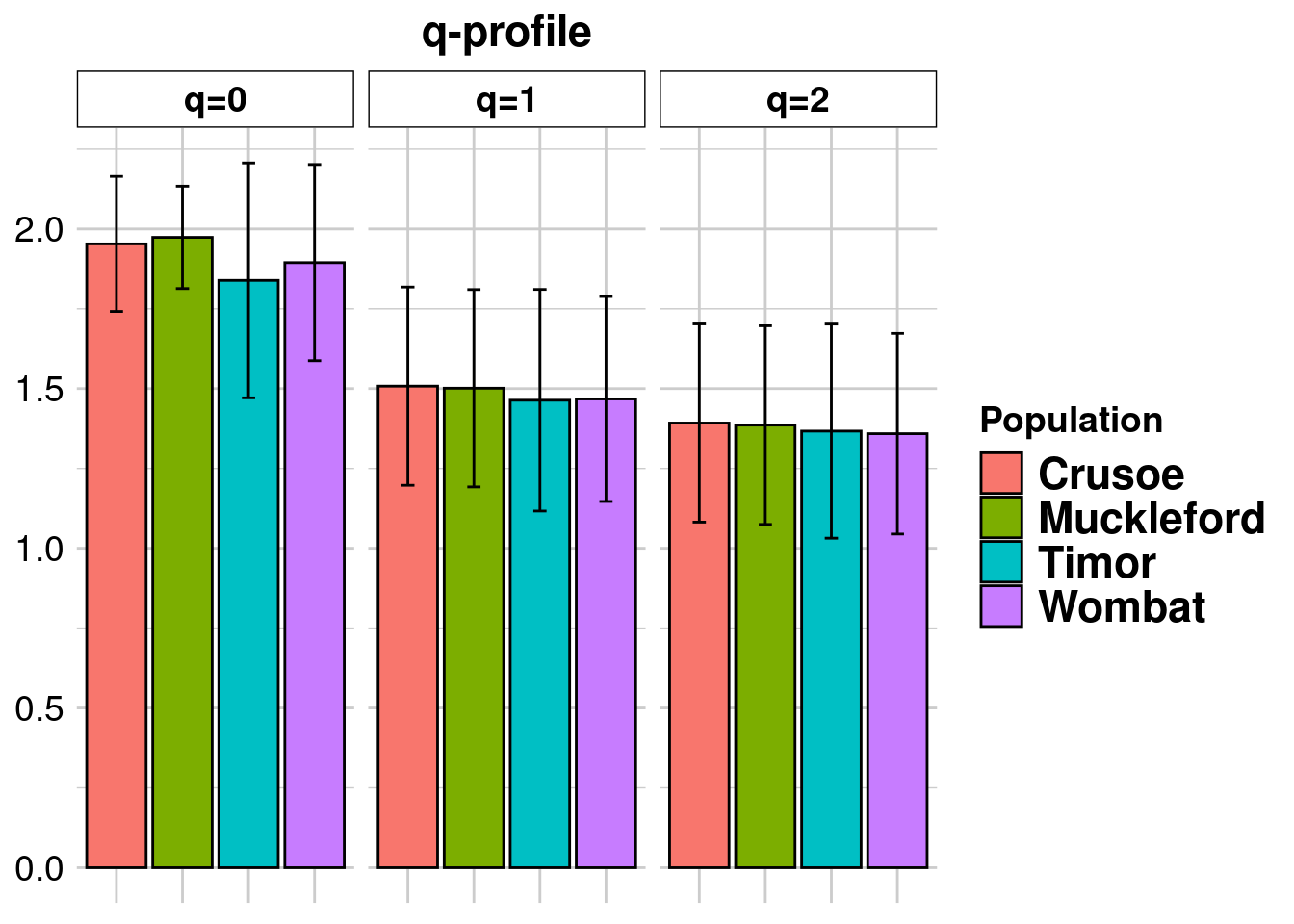
divers.correct <- dartR.base::gl.report.diversity(EYR.correct, pbar = FALSE, table = FALSE, verbose = 0) Processing genlight object with SNP data
divers.sloppy$one_H_alpha Crusoe Muckleford Timor Wombat
0.3884366 0.3844244 0.3519361 0.3591638 divers.correct$one_H_alpha Crusoe Muckleford Timor Wombat
0.3766483 0.3689045 0.3468530 0.3503540 divers.sloppy$one_H_beta Crusoe Muckleford Timor Wombat
Crusoe NA 0.02660219 0.09367676 0.06237102
Muckleford 0.006274769 NA 0.07326488 0.06577359
Timor 0.022518949 0.02452504 NA 0.08763552
Wombat 0.018905085 0.02091118 0.03715536 NAdivers.correct$one_H_beta Crusoe Muckleford Timor Wombat
Crusoe NA 0.02545762 0.08568969 0.05988791
Muckleford 0.005778848 NA 0.06967786 0.06243346
Timor 0.016804630 0.02067653 NA 0.08306746
Wombat 0.015054123 0.01892602 0.02995180 NA# Fixation indices
dartR.base::gl.fst.pop(EYR.sloppy, verbose = 0) Crusoe Muckleford Timor Wombat
Crusoe NA NA NA NA
Muckleford 0.03160198 NA NA NA
Timor 0.04023766 0.05408752 NA NA
Wombat 0.05466955 0.02407235 0.08452847 NAdartR.base::gl.fst.pop(EYR.correct, verbose = 0) Crusoe Muckleford Timor Wombat
Crusoe NA NA NA NA
Muckleford 0.02777612 NA NA NA
Timor 0.04008148 0.04786375 NA NA
Wombat 0.04413257 0.02208816 0.07111737 NAdartR.base::gl.report.fstat(EYR.sloppy, verbose = 0)Starting gl.colors
Selected color type div
Completed: gl.colors 
$Stat_matrices
$Stat_matrices$Fst
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0148 0.0171 0.0255
Muckleford 0.0148 NA 0.0249 0.0096
Timor 0.0171 0.0249 NA 0.0387
Wombat 0.0255 0.0096 0.0387 NA
$Stat_matrices$Fstp
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0356 0.0545 0.0669
Muckleford 0.0356 NA 0.0681 0.0337
Timor 0.0545 0.0681 NA 0.1046
Wombat 0.0669 0.0337 0.1046 NA
$Stat_matrices$Dest
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0236 0.0351 0.0434
Muckleford 0.0236 NA 0.0437 0.0213
Timor 0.0351 0.0437 NA 0.0657
Wombat 0.0434 0.0213 0.0657 NA
$Stat_matrices$Gst_H
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0566 0.0850 0.1047
Muckleford 0.0566 NA 0.1059 0.0525
Timor 0.0850 0.1059 NA 0.1603
Wombat 0.1047 0.0525 0.1603 NA
[[2]]
Stat_tables.Crusoe_vs_Muckleford Stat_tables.Crusoe_vs_Timor
Fst 0.0148 0.0171
Fstp 0.0356 0.0545
Dest 0.0236 0.0351
Gst_H 0.0566 0.0850
Stat_tables.Crusoe_vs_Wombat Stat_tables.Muckleford_vs_Timor
Fst 0.0255 0.0249
Fstp 0.0669 0.0681
Dest 0.0434 0.0437
Gst_H 0.1047 0.1059
Stat_tables.Muckleford_vs_Wombat Stat_tables.Timor_vs_Wombat
Fst 0.0096 0.0387
Fstp 0.0337 0.1046
Dest 0.0213 0.0657
Gst_H 0.0525 0.1603dartR.base::gl.report.fstat(EYR.correct, verbose = 0)Starting gl.colors
Selected color type div
Completed: gl.colors 
$Stat_matrices
$Stat_matrices$Fst
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0128 0.0168 0.0199
Muckleford 0.0128 NA 0.0212 0.0085
Timor 0.0168 0.0212 NA 0.0313
Wombat 0.0199 0.0085 0.0313 NA
$Stat_matrices$Fstp
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0317 0.0540 0.0556
Muckleford 0.0317 NA 0.0608 0.0315
Timor 0.0540 0.0608 NA 0.0903
Wombat 0.0556 0.0315 0.0903 NA
$Stat_matrices$Dest
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0199 0.0337 0.0344
Muckleford 0.0199 NA 0.0373 0.0189
Timor 0.0337 0.0373 NA 0.0547
Wombat 0.0344 0.0189 0.0547 NA
$Stat_matrices$Gst_H
Crusoe Muckleford Timor Wombat
Crusoe NA 0.0492 0.0832 0.0856
Muckleford 0.0492 NA 0.0930 0.0480
Timor 0.0832 0.0930 NA 0.1368
Wombat 0.0856 0.0480 0.1368 NA
[[2]]
Stat_tables.Crusoe_vs_Muckleford Stat_tables.Crusoe_vs_Timor
Fst 0.0128 0.0168
Fstp 0.0317 0.0540
Dest 0.0199 0.0337
Gst_H 0.0492 0.0832
Stat_tables.Crusoe_vs_Wombat Stat_tables.Muckleford_vs_Timor
Fst 0.0199 0.0212
Fstp 0.0556 0.0608
Dest 0.0344 0.0373
Gst_H 0.0856 0.0930
Stat_tables.Muckleford_vs_Wombat Stat_tables.Timor_vs_Wombat
Fst 0.0085 0.0313
Fstp 0.0315 0.0903
Dest 0.0189 0.0547
Gst_H 0.0480 0.1368Exercise data 3 - Bull shark
Data from Devloo‐Delva et al. (2023).
Load data
print(load("data/Bull_shark_DArTseq_genlight_for_sex-linked_markers.Rdata"))[1] "data.gl"data.gl@n.loc[1] 93202table(data.gl@pop)
E-ATL E-IO Fiji Japan N-IO W-ATL W-IO W-PAC
2 36 8 14 20 36 40 26 table(data.gl@other$ind.metrics$pop)
E-ATL E-IO Fiji Japan N-IO W-ATL W-IO W-PAC
2 36 8 14 20 36 40 26 table(data.gl@other$ind.metrics$sex, useNA = "ifany")
F M <NA>
85 64 33 1. Number of sex-linked markers?
ncores <- min(4, parallel::detectCores())
#still takes some minutes to run
res <- dartR.sexlinked::gl.filter.sexlinked(gl = data.gl, system = "xy", plots = TRUE, ncores = ncores)Detected 85 females and 64 males.Starting phase 1. Working in parallel...Building call rate plots.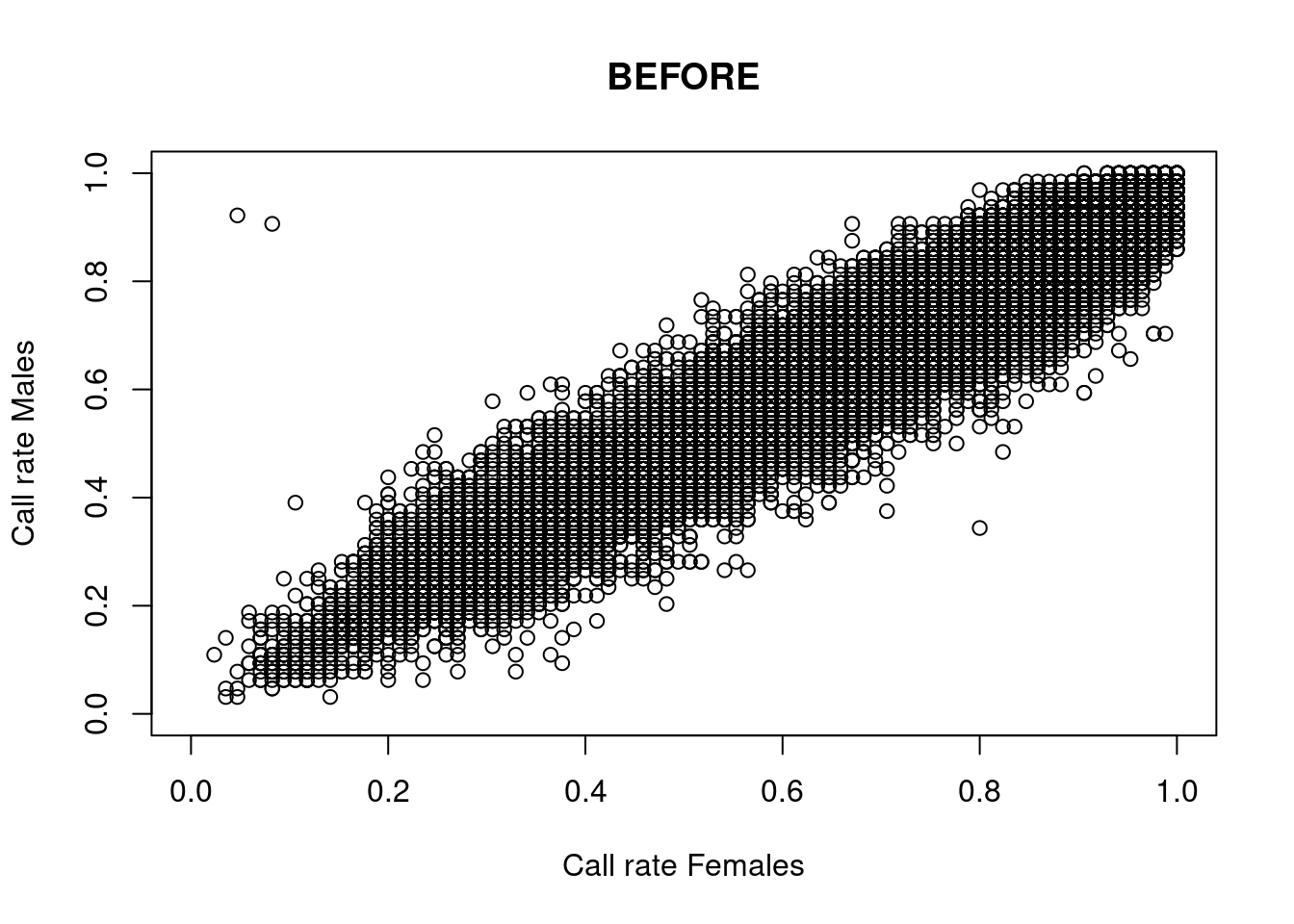
Done. Starting phase 2.Building heterozygosity plots.

Done building heterozygosity plots.**FINISHED** Total of analyzed loci: 93202.
Found 9 sex-linked loci:
2 Y-linked loci
2 sex-biased loci
4 X-linked loci
1 XY gametologs.
And 93193 autosomal loci.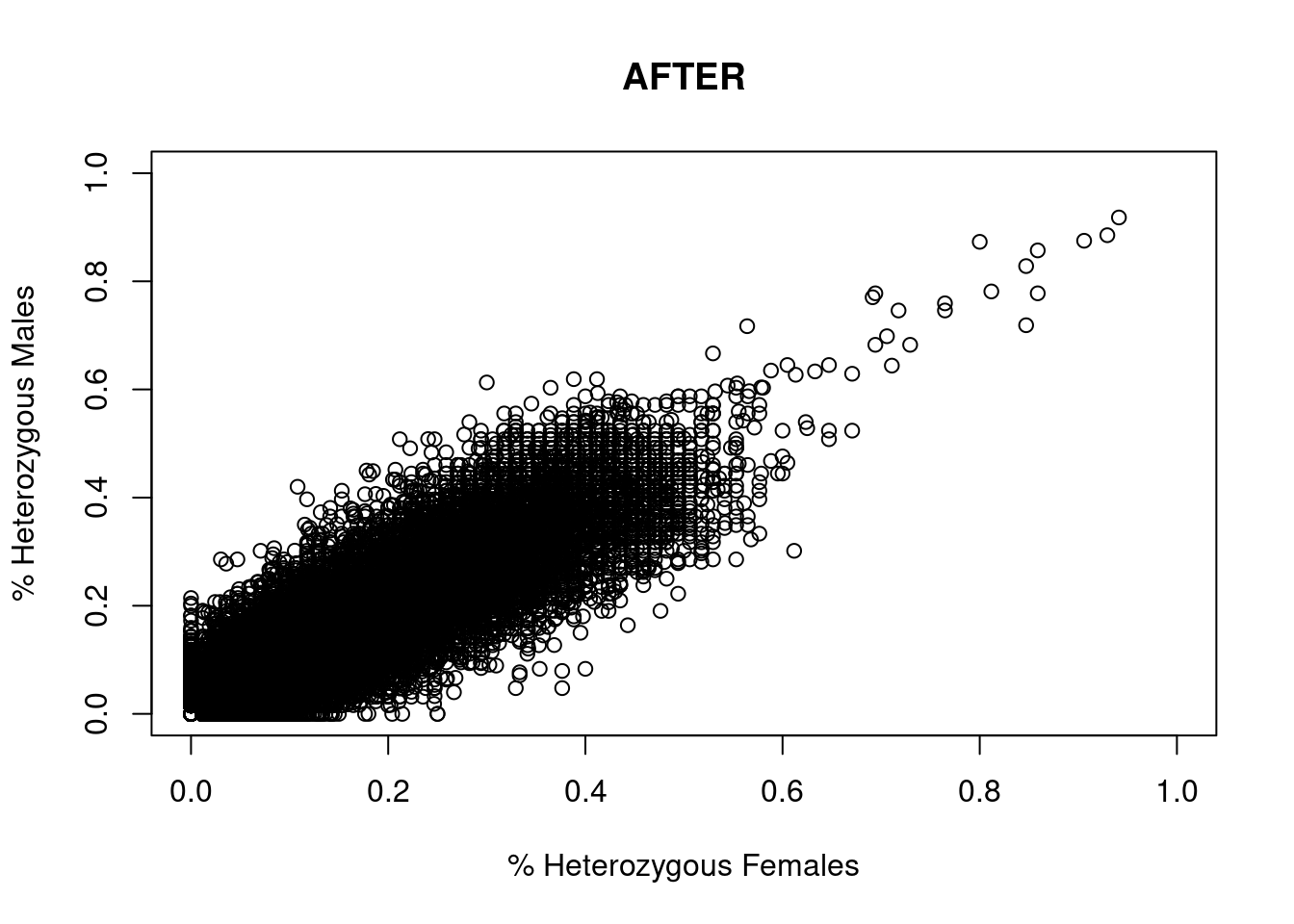
2. Individuals with wrong sexID?
#check if you can increase the number of cores
sexID <- dartR.sexlinked::gl.infer.sex(gl_sex_filtered = res, system = "xy", seed = 124)Not enough gametologs (need at least 5). Assigning NA...***FINISHED***knitr::kable(head(sexID))
agreed.sex <- sub(pattern = "\\*", replacement = "", x = sexID$agreed.sex) # remove asterisk
sum(data.gl$other$ind.metrics$sex != agreed.sex, na.rm = TRUE)| id | y.linked.sex | #called | #missing | x.linked.sex | #Het.x | #Hom.x | gametolog.sex | #Het.g | #Hom.g | agreed.sex | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CL-FIJ002-F | CL-FIJ002-F | F | 0 | 2 | F | 4 | 0 | NA | NA | NA | F |
| CL-FIJ003-M | CL-FIJ003-M | M | 2 | 0 | M | 0 | 4 | NA | NA | NA | M |
| CL-FIJ008-F | CL-FIJ008-F | F | 0 | 2 | F | 2 | 2 | NA | NA | NA | F |
| CL-FIJ010-F | CL-FIJ010-F | F | 0 | 2 | F | 3 | 1 | NA | NA | NA | F |
| CL-FIJ015-F | CL-FIJ015-F | F | 0 | 2 | F | 4 | 0 | NA | NA | NA | F |
| CL-FIJ018-F | CL-FIJ018-F | F | 0 | 2 | F | 3 | 1 | NA | NA | NA | F |
[1] 8Exercise data 4 - Blue shark
Data from Nikolic et al. (2023).
Load data
print(load("data/Blue_shark_DArTseq_genlight_for_sex-linked_markers.Rdata"))[1] "data.gl"data.gl@n.loc[1] 172384table(data.gl@pop)
EIO MED1 MED2 NATL NEATL NIO NPAC SAF1 SAF2 SWPAC1 SWPAC2
8 34 20 49 26 27 4 21 89 30 16
SWPAC3 WIO
11 29 table(data.gl@other$ind.metrics$pop)
EIO MED1 MED2 NATL NEATL NIO NPAC SAF1 SAF2 SWPAC1 SWPAC2
8 34 20 49 26 27 4 21 89 30 16
SWPAC3 WIO
11 29 table(data.gl@other$ind.metrics$sex, useNA = "ifany")
F M <NA>
104 111 149 1. Number of sex-linked markers?
#check if you can increase the number of cores
# ncores <- min(4,parallel::detectCores())
# resbull <- dartR.sexlinked::gl.filter.sexlinked(gl = data.gl, system = "xy", plots = TRUE, ncores = ncores)
#load results from previous run
resbull <- readRDS("./data/resbull.rds")2. Individuals with wrong sexID?
#check if you can increase the number of cores
sexID <- dartR.sexlinked::gl.infer.sex(gl_sex_filtered = resbull, system = "xy", seed = 124)Not enough gametologs (need at least 5). Assigning NA...***FINISHED***knitr::kable(head(sexID))
agreed.sex <- sub(pattern = "\\*", replacement = "", x = sexID$agreed.sex) # remove asterisk
sum(data.gl$other$ind.metrics$sex != agreed.sex, na.rm = TRUE)| id | y.linked.sex | #called | #missing | x.linked.sex | #Het.x | #Hom.x | gametolog.sex | #Het.g | #Hom.g | agreed.sex | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 60088 | 60088 | M | 2 | 0 | M | 0 | 22 | NA | NA | NA | M |
| 60160 | 60160 | M | 2 | 0 | M | 0 | 21 | NA | NA | NA | M |
| 60168 | 60168 | M | 2 | 0 | M | 0 | 22 | NA | NA | NA | M |
| 60176 | 60176 | M | 2 | 0 | M | 0 | 21 | NA | NA | NA | M |
| 60096 | 60096 | M | 2 | 0 | M | 0 | 22 | NA | NA | NA | M |
| 60104 | 60104 | M | 2 | 0 | M | 1 | 19 | NA | NA | NA | M |
[1] 22